Efekt farmakologiczny. Fizykochemiczne i chemiczne mechanizmy działania substancji leczniczych. Zindywidualizowana strategia terapii lekowej
Bloker receptora histaminowego H 1 pierwszej generacji. Wpływ na ośrodkowy układ nerwowy wynika z blokady receptorów histaminowych H3 w mózgu i zahamowania ośrodkowych struktur cholinergicznych. Łagodzi skurcze mięśni gładkich (działanie bezpośrednie), zmniejsza przepuszczalność naczyń włosowatych, zapobiega i osłabia reakcje alergiczne, działa miejscowo znieczulająco, przeciwwymiotnie, uspokajająco, umiarkowanie blokuje receptory cholinergiczne zwojów autonomicznych, działa nasennie.
Wskazania - reakcje anafilaktyczne i rzekomoanafilaktyczne (w terapii skojarzonej) - obrzęk Quinckego;
Choroba posurowicza, - inne ostre stany alergiczne (w terapii skojarzonej oraz w przypadkach, gdy stosowanie tabletek jest niemożliwe).
Skutki uboczne
Z boku system nerwowy: senność, osłabienie, zmniejszenie szybkości reakcji psychomotorycznej, zaburzenia koordynacji ruchów, zawroty głowy, drżenie, drażliwość, euforia, pobudzenie (szczególnie u dzieci), bezsenność.
Z boku Układ oddechowy: suchość błony śluzowej jamy ustnej, nosa, oskrzeli (zwiększona lepkość plwociny).
Od strony hematopoezy: niedokrwistość hemolityczna, małopłytkowość, agranulocytoza.
Z boku układu sercowo-naczyniowego: spadek ciśnienia krwi, tachykardia, skurcz dodatkowy.
Z układu moczowego: zaburzenia oddawania moczu.
Reakcje alergiczne: pokrzywka, nadwrażliwość na światło, wysypka skórna, swędzenie.
Loratadyna (klarytyna)
efekt farmakologiczny
Lek przeciwalergiczny, selektywny bloker obwodowych receptorów histaminowych H1. Loratadyna jest związkiem trójpierścieniowym o wyraźnym działaniu przeciwhistaminowym. Ma szybkie i długotrwałe działanie antyalergiczne.
Loratadyna nie przenika do BBB i nie wpływa na centralny układ nerwowy. Nie ma klinicznie istotnego działania antycholinergicznego ani uspokajającego, tj. nie powoduje senności i nie wpływa na szybkość reakcji psychomotorycznych przy stosowaniu w zalecanych dawkach. Przyjmowanie Claritin nie prowadzi do wydłużenia odstępu QT w EKG. Na leczenie długoterminowe nieobserwowane klinicznie znaczące zmiany parametry życiowe, dane z badań fizykalnych, wyniki laboratoryjne lub EKG.
Loratadyna nie wykazuje znaczącej selektywności wobec receptorów histaminowych H2. Nie hamuje wychwytu zwrotnego noradrenaliny i ma niewielki lub żaden wpływ na układ sercowo-naczyniowy lub funkcję rozrusznika
Skutki uboczne
Z układu nerwowego: u dzieci w wieku od 2 do 12 lat - ból głowy (2,7%), nerwowość (2,3%), zmęczenie (1%); u dorosłych - bół głowy(0,6%), senność (1,2%), bezsenność (0,1%).
u dorosłych - zwiększony apetyt (0,5%).
W okresie po wprowadzeniu do obrotu
Z układu nerwowego: rzadko (< 1/10 000) - головокружение, утомляемость.
Z boku układ trawienny: rzadko (< 1/10 000) - сухость во рту, желудочно-кишечные расстройства (тошнота, гастрит), нарушение функции печени.
Reakcje alergiczne: rzadko (< 1/10 000) - сыпь, анафилаксия.
Ze strony układu sercowo-naczyniowego: rzadko (< 1/10 000) - сердцебиение, тахикардия.
Z boku skóra: rzadko (< 1/10 000) - алопеция.
Wskazania
sezonowy (katar sienny) i całoroczny alergiczny nieżyt nosa i alergiczne zapalenie spojówek (w celu wyeliminowania objawów związanych z tymi chorobami - kichanie, swędzenie błony śluzowej nosa, wyciek z nosa, pieczenie i swędzenie oczu, łzawienie); - przewlekła pokrzywka idiopatyczna; choroby skóry pochodzenia alergicznego ...
33. Środki eliminujące powszechne przejawy reakcje alergiczne rodzaj wstrząsu anafilaktycznego. Epinefryna, Eufillin, prednizolon. Główne efekty farmakologiczne, cel przepisywania każdego leku i działania niepożądane.
NARKOTYKI ANTYALERGICZNE(przeciwalergiczne; syn. środki odczulające) - leki, które zapobiegają lub osłabiają objawy reakcji alergicznych. Jeden z możliwe sposoby zapobieganie i leczenie reakcji alergicznych to tzw. metoda. specyficzne odczulenie, czyli obniżenie wrażliwości organizmu na dowolny antygen poprzez wielokrotne podawanie samego antygenu w małych dawkach, które nie powodują objawów alergii. W takim przypadku organizm stopniowo traci wrażliwość na wstrzyknięty antygen.
Adrenalina
Działanie farmakologiczne Adrenomimetyk, działa bezpośrednio stymulująco na receptory α- i β-adrenergiczne. Pod wpływem epinefryny (adrenaliny) w wyniku pobudzenia receptorów α-adrenergicznych dochodzi do wzrostu zawartości wapnia wewnątrzkomórkowego w mięśniach gładkich. zwiększa aktywność fosfolipazy C (poprzez stymulację białka G) oraz tworzenie trifosforanu inozytolu i diacyloglicerolu. Ma wyraźny wpływ na układ sercowo-naczyniowy. Zwiększa tętno i siłę, udar i rzut serca. Poprawia przewodność AV, zwiększa automatyzm. Zwiększa zapotrzebowanie mięśnia sercowego na tlen. Powoduje zwężenie naczyń narządów jamy brzusznej, skóry, błon śluzowych, w mniejszym stopniu - mięśni szkieletowych. Zwiększa ciśnienie krwi (głównie skurczowe), Epinefryna (adrenalina) rozluźnia mięśnie gładkie oskrzeli, obniża napięcie i motorykę przewodu pokarmowego, rozszerza źrenice, pomaga obniżyć ciśnienie wewnątrzgałkowe. Powoduje hiperglikemię i zwiększa ilość wolnych kwasów tłuszczowych w osoczu. Wskazania Reakcje alergiczne typu natychmiastowego (m.in. pokrzywka, obrzęk naczynioruchowy, wstrząs anafilaktyczny), rozwijające się przy stosowaniu leków, surowic, transfuzji krwi, pokarmów, ukąszeń owadów lub innych alergenów. Astma oskrzelowa (łagodzenie ataku), skurcz oskrzeli podczas znieczulenia. Aby zatrzymać krwawienie. Skutek uboczny Od strony układu sercowo-naczyniowego: dławica piersiowa, bradykardia lub tachykardia, kołatanie serca, wzrost lub spadek ciśnienia krwi; przy stosowaniu w dużych dawkach - komorowe zaburzenia rytmu; rzadko - arytmia, ból w klatce piersiowej. Z układu nerwowego: ból głowy, lęk, drżenie, zawroty głowy, nerwowość, zmęczenie, zaburzenia psychonerwicowe
Eufilina
efekt farmakologiczny
lek rozszerzający oskrzela, pochodna ksantyny; hamuje fosfodiesterazę, zwiększa akumulację cyklicznego adenozynomonofosforanu w tkankach, blokuje receptory adenozyny (puryny); ogranicza przepływ jonów wapnia przez kanały błon komórkowych, zmniejsza aktywność skurczową mięśni gładkich. Rozluźnia mięśnie oskrzeli, zwiększa klirens śluzowo-rzęskowy, stymuluje skurcz przepony, poprawia pracę mięśni oddechowych i międzyżebrowych, stymuluje ośrodek oddechowy, zwiększa jego wrażliwość na dwutlenek węgla i poprawia wentylację pęcherzykową, co ostatecznie prowadzi do zmniejszenia w nasileniu i częstotliwości epizodów bezdechu. Normalizując czynność oddechową przyczynia się do nasycenia krwi tlenem oraz zmniejszenia stężenia dwutlenku węgla. Działa pobudzająco na czynność serca, zwiększa siłę i liczbę skurczów serca, zwiększa przepływ wieńcowy i zapotrzebowanie mięśnia sercowego na tlen. Zmniejsza napięcie naczyń krwionośnych (głównie mózgu, skóry i nerek). Działa rozszerzająco na żyły obwodowe, zmniejsza opór naczyń płucnych, obniża ciśnienie w „małym” kręgu krążenia krwi. Zwiększa przepływ krwi przez nerki, ma umiarkowane działanie moczopędne. Rozszerza pozawątrobowe drogi żółciowe. Hamuje agregację płytek krwi (hamuje czynnik aktywacji płytek i PgE2 alfa), zwiększa odporność erytrocytów na deformację (poprawia właściwości reologiczne krwi), ogranicza tworzenie skrzepliny i normalizuje mikrokrążenie. Ma działanie tokolityczne, zwiększa kwasowość soku żołądkowego. Stosowany w dużych dawkach ma działanie elileptogenne.
Skutki uboczne
Z układu nerwowego: zawroty głowy, ból głowy, bezsenność, pobudzenie, niepokój, drażliwość, drżenie.
Ze strony układu sercowo-naczyniowego: kołatanie serca, tachykardia (w tym u płodu podczas przyjmowania kobiety w ciąży w trzecim trymestrze), zaburzenia rytmu serca, bóle serca, obniżenie ciśnienia krwi, zwiększenie częstości napadów dusznicy bolesnej.
Z układu pokarmowego: ból żołądka, nudności, wymioty, refluks żołądkowo-przełykowy, zgaga, zaostrzenie choroby wrzodowej, biegunka, przy długotrwałym stosowaniu - zmniejszenie apetytu.
Reakcje alergiczne: wysypka skórna, swędzenie, gorączka.
Inni: ból w klatce piersiowej, przyspieszony oddech, uderzenia gorąca, albuminuria, krwiomocz, hipoglikemia, zwiększone wydalanie moczu, zwiększona potliwość.
Wskazania
Zespół obturacyjny oskrzeli o dowolnej genezie: astma oskrzelowa (lek z wyboru u chorych na astmę wysiłkową oraz jako lek dodatkowy przy innych postaciach), przewlekła obturacyjna choroba płuc, rozedma płuc, przewlekłe obturacyjne zapalenie oskrzeli, nadciśnienie płucne, serce płucne, sen bezdech.
Prednizolon
efekt farmakologiczny
Syntetyczny GCS. Ma wyraźne działanie przeciwzapalne. Lek hamuje rozwój objawów zapalenia. Hamuje akumulację makrofagów, leukocytów i innych komórek w obszarze stanu zapalnego. Hamuje fagocytozę, uwalnianie enzymów mikrosomalnych oraz syntezę i uwalnianie mediatorów zapalnych. Powoduje zmniejszenie przepuszczalności naczyń włosowatych, zahamowanie migracji leukocytów.
Nasila syntezę lipomoduliny, inhibitora fosfolipazy A2, która uwalnia kwas arachidonowy z błon fosfolipidowych, jednocześnie hamując jego syntezę.
Mechanizm immunosupresyjnego działania prednizonu nie jest w pełni poznany. Lek zmniejsza liczbę limfocytów T, monocytów i granulocytów kwasochłonnych, a także wiązanie immunoglobulin z receptorami na powierzchni komórki, hamuje syntezę lub uwalnianie interleukin poprzez zmniejszenie blastogenezy limfocytów T; zmniejsza wczesną odpowiedź immunologiczną. Hamuje również przenikanie kompleksów immunologicznych przez błony oraz zmniejsza stężenie składników dopełniacza i immunoglobulin.
Prednizolon działa na dystalne kanaliki nerkowe, zwiększając wchłanianie zwrotne sodu i wody, a także zwiększając wydalanie jonów potasu i jonów wodorowych.
Prednizolon hamuje wydzielanie ACTH przez przysadkę mózgową, co prowadzi do zmniejszenia produkcji kortykosteroidów i androgenów przez korę nadnerczy. Po długotrwałym stosowaniu leku w dużych dawkach funkcja nadnerczy może zostać przywrócona w ciągu roku, aw niektórych przypadkach rozwija się uporczywe tłumienie ich funkcji. Prednizolon nasila katabolizm białek i indukuje enzymy biorące udział w metabolizmie aminokwasów. Hamuje syntezę i wzmaga katabolizm białek w tkance limfatycznej, łącznej, mięśniowej. Przy długotrwałym stosowaniu możliwy jest rozwój atrofii tych tkanek (jak również skóry).
Zwiększa stężenie glukozy we krwi poprzez indukcję enzymów glukoneogenezy w wątrobie, stymulację katabolizmu białek (co zwiększa ilość aminokwasów do glukoneogenezy) oraz zmniejszenie zużycia glukozy w tkankach obwodowych. Prowadzi to do nagromadzenia glikogenu w wątrobie, wzrostu stężenia glukozy we krwi i wzrostu insulinooporności.
Wskazania
Choroby endokrynologiczne:
Niewydolność kory nadnerczy: pierwotna (choroba Addisona) i wtórna; - zespół adrenogenitalny (wrodzony przerost nadnerczy); - ostra niewydolność kory nadnerczy;
Przed interwencjami chirurgicznymi oraz w przypadku poważnych chorób i urazów u pacjentów z niedoczynnością kory nadnerczy; - podostre zapalenie tarczycy.
Ciężkie choroby alergiczne oporne na inne terapie: - kontaktowe zapalenie skóry; - atopowe zapalenie skóry; - choroba posurowicza; - reakcje nadwrażliwości na leki;
Stałe lub sezonowe alergiczny nieżyt nosa; - reakcje anafilaktyczne; - obrzęk naczynioruchowy.
Choroby reumatyczne:
Reumatoidalne zapalenie stawów, młodzieńcze reumatoidalne zapalenie stawów (w przypadkach opornych na inne leczenie);
Choroby dermatologiczne: - złuszczające zapalenie skóry, - opryszczkowate pęcherzowe zapalenie skóry;
Ciężkie łojotokowe zapalenie skóry, - Ciężki rumień wielopostaciowy (zespół Stevensa-Johnsona);
Skutki uboczne
Przy krótkotrwałym stosowaniu prednizolonu (podobnie jak innych kortykosteroidów) działania niepożądane występują rzadko. Podczas długotrwałego stosowania prednizolonu mogą wystąpić następujące działania niepożądane.
Ze strony bilansu wodno-elektrolitowego: zatrzymanie sodu i płynów w organizmie, hipokaliemia.
Z układu mięśniowo-szkieletowego: osłabienie mięśni, miopatia steroidowa, utrata masy mięśniowej, osteoporoza, kompresyjne złamanie kręgosłupa.
Z układu pokarmowego: wrzód steroidowy z możliwą perforacją i krwawieniem, zapalenie trzustki, wzdęcia, wrzodziejące zapalenie przełyku, niestrawność, nudności, zwiększony apetyt.
Reakcje dermatologiczne: zanik skóry, rozstępy, trądzik, opóźnione gojenie się ran, ścieńczenie skóry, wybroczyny, krwiaki, rumień, wzmożone pocenie się, alergiczne zapalenie skóry, pokrzywka, obrzęk naczynioruchowy.
Od strony ośrodkowego układu nerwowego i obwodowego układu nerwowego: zwiększone ciśnienie śródczaszkowe z zespołem zastoinowej brodawki nerwu wzrokowego (występuje najczęściej u dzieci, po zbyt szybkim zmniejszeniu dawki, objawy - ból głowy, pogorszenie ostrości wzroku, podwójne widzenie); drgawki, zawroty głowy, ból głowy, zaburzenia snu.
Ze stanu hormonalnego: wtórna niewydolność nadnerczy i podwzgórze-przysadka (szczególnie w sytuacjach stresowych: choroba, uraz, operacja); Zespół Cushinga.
Inni: reakcje anafilaktyczne, reakcje nadwrażliwości; zarostowe zapalenie tętnic, przyrost masy ciała, omdlenia.
1. Istota farmakologii jako nauki. Działy i dziedziny współczesnej farmakologii. Podstawowe pojęcia i pojęcia farmakologii – działanie farmakologiczne, działanie, skuteczność, substancje chemiczne.
Farmakologia- nauka o lekach we wszystkich aspektach - podstawy teoretyczne terapia:
a) nauka o interakcji chemikaliów z żywymi systemami
b) nauka o zarządzaniu procesami życiowymi organizmu za pomocą chemikaliów
Rozwój farmakologii idzie w dwóch głównych kierunkach: podstawowe badania wyjaśnienie zasad i mechanizmów działania leków i opracowanie skuteczne leki jako podstawa leczenia chorób.
Farmakologia dzieli się na:
1. Generał- bada ogólne wzorce interakcji substancje lecznicze z żywymi organizmami.
Prywatny- uwzględnia określone grupy farmakologiczne i poszczególne leki
2. Eksperymentalna (podstawowa) farmakologia- bada działanie leków w eksperymencie.
Farmakologia kliniczna- bada skuteczność kliniczną i bezpieczeństwo
stosowanie leków u pacjentów, optymalizuje program leczenia pacjenta z uwzględnieniem
jego stan.
Toksykologia- bada toksyczny wpływ na narządy różne substancje(włącznie z
i lecznicze).
Działy współczesnej farmakologii:
1) farmakodynamika- badania a) wpływ leków na organizm człowieka, b) interakcje różnych leków w organizmie podczas ich przepisywania, c) wpływ wieku i różne choroby na wpływ leków
2) farmakokinetyka- bada wchłanianie, dystrybucję, metabolizm i wydalanie leków (czyli jak organizm pacjenta reaguje na leki)
3) farmakogenetyka- bada rolę czynników genetycznych w kształtowaniu odpowiedzi farmakologicznej organizmu na leki
4) farmakoekonomika- ocenia efekty stosowania i koszt leków w celu podjęcia decyzji o ich późniejszym praktycznym zastosowaniu
5) farmakoepidemiologia- bada zażywanie narkotyków i ich skutki na poziomie populacji, lub duże grupy ludzi, aby zapewnić stosowanie najskuteczniejszych i najbezpieczniejszych leków
Podstawowe terminy i pojęcia:
Aktywność farmakologiczna (biologiczna)- właściwość substancji do wywoływania zmian w biosystemie (ciało ludzkie). Substancje farmakologiczne = biologicznie substancje aktywne(BAS)
efekt farmakologiczny- wpływ narkotyków na obiekt i jego cele
Efekt farmakologiczny- wynik działania substancji w organizmie (modyfikacja procesów fizjologicznych, biochemicznych, struktur morfologicznych) - ilościowy, ale nie zmiana jakościowa w stanie biosystemów (komórek, tkanek, narządów).
Skuteczność leków- zdolność leków do wywoływania pewnych niezbędnych ta sprawa efekty farmakologiczne w organizmie. Oceniany na podstawie „istotnych dowodów” – adekwatnych, dobrze kontrolowanych badań i badań klinicznych prowadzonych przez ekspertów z odpowiednim wykształceniem naukowym i doświadczeniem w badaniach leków tego typu (FDA)
2. Źródła i etapy powstawania leków. Leki - leki generyczne, placebo - efekty Definicja pojęć lek, lek, lek i postać dawkowania.
Źródła powstawania leków:
a) surowce naturalne: rośliny, zwierzęta, minerały, produkty odpadowe mikroorganizmów (glikozydy nasercowe, insulina wieprzowa, AB)
b) modyfikowane naturalne substancje biologicznie czynne
c) produkty syntezy chemicznej (metody: skrining farmakologiczny, projektowanie molekularne, reprodukcja amin biogennych, celowana modyfikacja cząsteczek o znanej już aktywności, synteza farmakologicznie czynnych metabolitów, wyniki losowe (metoda „serendypityczna”)
d) produkty inżynierii genetycznej (rekombinowana insulina, interferony)
Etapy tworzenia leku:
1. Synteza leków w laboratorium chemicznym
2. Ocena przedkliniczna działania i działań niepożądanych leków Ministerstwa Zdrowia i innych organizmów
3. Badania kliniczne leków Badanie dokumentacji przez Komisję Farmakologiczną przeprowadza się po zakończeniu każdej fazy. Lek można wycofać na dowolnym etapie. (I faza – ocena tolerancji u zdrowych ochotników w wieku 20-25 lat, II faza – u chorych ochotników poniżej 100 osób cierpiących na określoną chorobę, III faza – wieloośrodkowe badania kliniczne na dużych grupach osób (do 1000 osób) IV faza - monitorowanie leku w ciągu 5 lat od jego oficjalnej zgody (wykonywane na dużej liczbie pacjentów (co najmniej 1000-5000 osób). Po zakończeniu III fazy badań klinicznych dokumentacja ponownie trafia do Komisji Farmakologicznej ( objętość pełnej dokumentacji może wynosić do 1 miliona stron) i w ciągu 1-2 lat zarejestrowana w Państwowym Rejestrze Leków i Produktów cel medyczny... Dopiero po tym koncern farmaceutyczny ma prawo rozpocząć produkcję przemysłową produkt leczniczy i jego dystrybucja za pośrednictwem sieci aptek.
Lek generyczny to lek generyczny będący repliką leku oryginalnego, na który wygasł patent. Może różnić się od oryginalnego leku składem zaróbek. Warunkiem sprzedaży leków generycznych jest poparta dowodami równoważność farmaceutyczna, biologiczna i terapeutyczna z lekiem macierzystym. Leki generyczne są zawsze tańsze niż ich markowe odpowiedniki, ponieważ firma nie wydaje pieniędzy na 10-15 lat badań nad lekami, ale korzysta z gotowych danych
Placebo- jakikolwiek składnik terapii, który nie ma żadnego specyficznego biologicznego wpływu na leczoną chorobę.
Jest używany w celu kontroli podczas oceny działania leków oraz w celu zapewnienia pacjentowi korzyści bez żadnych środki farmakologiczne w wyniku jedynie wpływu psychologicznego (tj. efekt placebo).
Wszystkie zabiegi mają komponent psychologiczny lub satysfakcjonujące ( efekt placebo), lub kłopotliwe (efekt nocebo). Przykład efektu placebo: szybka poprawa u pacjenta Infekcja wirusowa podczas stosowania antybiotyków. Korzyść z efektu placebo jest związana z wpływ psychologiczny na pacjenta. Będzie maksymalny tylko podczas korzystania z niego. w połączeniu z metodami leczenia które mają wyraźny specyficzny efekt. Drogie substancje jako placebo również pomagają osiągnąć większą odpowiedź.
Wskazania do stosowania placebo:
1) słaby zaburzenia psychiczne
2) wsparcie psychologiczne pacjent z nieuleczalną choroba przewlekła lub z podejrzeniem poważnej diagnozy
Medycyna- wszelkie substancje lub produkty stosowane do modyfikowania lub badania układów fizjologicznych lub stanów patologicznych z korzyścią dla biorcy (zgodnie z WHO, 1966); poszczególne substancje, mieszaniny substancji lub kompozycje o nieznanym składzie o potwierdzonych właściwościach leczniczych.
Substancja lecznicza- indywidualny związek chemiczny stosowany jako lek.
Forma dawkowania- wygodny dla praktyczne zastosowanie forma podawana lekowi w celu uzyskania pożądanego efektu terapeutycznego lub profilaktycznego.
Produkt leczniczy- produkt leczniczy w określonej postaci dawkowania, zatwierdzony przez organ rządowy.
Na przykład: lekiem jest antybiotyk ampicylina, lekiem jest trójwodzian ampicyliny, który może mieć postać tabletek lub kapsułek. Lekem są tabletki trihydratu ampicyliny po 0,25 g każda.
Drogi podawania leków do organizmu i ich charakterystyka. Przedsystemowa eliminacja leków.
ale. dojelitowa droga podania: doustna, podjęzykowa, policzkowa, doodbytnicza, rurkowa b. pozajelitowa droga podania: dożylnie, podskórnie, domięśniowo, ... 2. W przypadku narażenia miejscowego: skóra (naskórkowa), na błonach śluzowych, w jamach (brzucha, opłucnej, stawu), w tkance ...Transport leków przez bariery biologiczne i jego odmiany. Główne czynniki wpływające na transport leków w organizmie.
1) Filtracja (dyfuzja wody) - pasywny ruch cząsteczek substancji wzdłuż gradientu stężenia przez pory wypełnione wodą w błonie każdego ... 2) Dyfuzja pasywna (dyfuzja lipidów) - główny mechanizm przenoszenia leków, ... 3 ) Transport za pomocą określonych nośników – transfer leków za pomocą nośników wbudowanych w membranę (częściej…Transport przez błony substancji leczniczych o zmiennej jonizacji (równanie jonizacji Hendersona-Hasselbalcha). Zasady kontroli transferu.
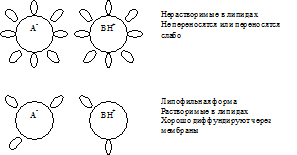
Wszystkie leki są słabymi kwasami lub słabymi zasadami, które mają własne wartości stałej jonizacji (pK). Jeżeli wartość pH podłoża jest równa wartości pK leku, to 50% jego cząsteczek będzie w stanie zjonizowanym, a 50% w stanie niezjonizowanym, a podłoże dla leku będzie obojętne.
W środowisko kwaśne(pH jest mniejsze od pK), przy nadmiarze protonów słaby kwas będzie w postaci niezdysocjowanej (R-COOH), czyli będzie związany z protonem - protonowany. Ta forma kwasu jest nienaładowana i łatwo rozpuszczalna w lipidach. Jeśli pH zostanie przesunięte na stronę zasadową (tj. pH stanie się większe niż pK), kwas zacznie dysocjować i traci proton, przechodząc do postaci nieprotonowanej, która ma ładunek i jest słabo rozpuszczalna w lipidach.
W środowisku alkalicznym, gdzie występuje niedobór protonów, słaba zasada będzie w formie niezdysocjowanej (R-NH 2), tj. będzie nieprotonowany i pozbawiony ładunku. Ta forma bazy jest dobrze rozpuszczalna w tłuszczach i szybko wchłaniana. W środowisku kwaśnym występuje nadmiar protonów i słaba zasada zacznie się dysocjować, wiążąc protony i tworząc protonowaną, naładowaną formę zasady. Ta forma jest słabo rozpuszczalna w lipidach i słabo wchłaniana.
W konsekwencji, wchłanianie słabych kwasów następuje głównie w środowisku kwaśnym, a słabych zasad w środowisku zasadowym.
Cechy metabolizmu słabych kwasów (SC):
1) żołądek: SA w kwaśnej treści żołądka nie ulega jonizacji, aw zasadowym środowisku jelita cienkiego ulega dysocjacji i cząsteczki SA nabierają ładunku. Dlatego wchłanianie słabych kwasów będzie najintensywniejsze w żołądku.
2) we krwi podłoże jest wystarczająco alkaliczne, a wchłonięte cząsteczki SC przekształcą się w formę zjonizowaną. Filtr kłębuszków nerek umożliwia przechodzenie zarówno zjonizowanych, jak i niezjonizowanych cząsteczek, dlatego pomimo ładunku cząsteczki, SC zostaną wydalone do mocz pierwotny
3) jeśli mocz ma odczyn zasadowy, kwas pozostanie w formie zjonizowanej, nie będzie mógł ponownie wchłonąć się do krwiobiegu i zostanie wydalony z moczem; Jeśli mocz jest kwaśny, lek przyjmie postać niezjonizowaną, która jest łatwo wchłaniana z powrotem do krwi.
Cechy metabolizmu słabych zasad: przeciwnie do SC (lepsze wchłanianie w jelicie; w moczu zasadowym są ponownie wchłaniane)
To., aby przyspieszyć usuwanie słabego kwasu z organizmu, mocz musi być alkalizowany, a aby przyspieszyć usuwanie słabej zasady, musi być zakwaszony (detoksykacja wg Popowa).
Ilościowa zależność procesu jonizacji leku przy różnym pH środowiska pozwala na otrzymanie równania Henderson-Hasselbach:
Gdzie pKa odpowiada wartości pH, przy której stężenia postaci zjonizowanej i niezjonizowanej są w równowadze .
Równanie Hendersona-Hasselbacha pozwala oszacować stopień jonizacji leku przy danej wartości pH oraz przewidzieć prawdopodobieństwo jego przeniknięcia przez błonę komórkową.
(1)Dla rozcieńczonego kwasu A,
HA ↔ H + + A -, gdzie HA jest stężeniem niezjonizowanej (protonowanej) formy kwasu, a A - jest stężeniem formy zjonizowanej (nieprotonowanej).
(2) Dla słaba podstawa, B,
BH + ↔ H + + B, gdzie BH + to stężenie protonowanej formy zasady, B to stężenie formy nieprotonowanej
Znając pH pożywki i pKa substancji, z obliczonego logarytmu można wyznaczyć stopień jonizacji leku, a tym samym stopień jego wchłaniania z przewód pokarmowy, reabsorpcja lub wydalanie przez nerki z różne znaczenia pH moczu itp.
Zasady kontroli transportu.
Aby przyspieszyć wchłanianie
Ø słabe kwasy (ASA) - pH soku żołądkowego powinno być kwaśne;
Ø słaba baza - pH soku żołądkowego powinno być neutralne.
Aby przyspieszyć wypłatę
Ø słabe kwasy - zalkalizowany mocz;
Ø słabe zasady - zakwaszają mocz.
Przenoszenie leków w ciele. Dyfuzja wody i dyfuzja w lipidach (prawo Ficka). Transport aktywny.
Przenoszenie leków w organizmie może odbywać się poprzez dyfuzję wody i lipidów, transport aktywny, endocytozę i pinocytozę.
Cechy przenoszenia leków w organizmie przez dyfuzję wody:
1. Powłoki nabłonkowe (błony śluzowe przewodu pokarmowego, jamy ustnej itp.) - dyfuzja wody tylko bardzo małych cząsteczek (metanol, jony litu itp.)
2. Kapilary (z wyjątkiem mózgowych) - filtracja substancji o masie cząsteczkowej do 20-30 tysięcy Tak.
3. Naczynia włosowate mózgu - w zasadzie nie posiadają porów wodnych, z wyjątkiem okolic przysadki mózgowej, szyszynki, komory IV strefy, splotu naczyniówkowego, wzniosłości środkowej
4. Łożysko - nie ma porów wodnych (choć kontrowersyjny problem).
5. Wiązanie leków z białkami krwi zapobiega ich uwalnianiu z krwiobiegu, a tym samym dyfuzji wody
6. Dyfuzja w wodzie zależy od wielkości cząsteczek leku i porów wodnych
Cechy dyfuzji lipidów:
1. Główny mechanizm przenoszenia leku przez błony komórkowe
2. Wyznaczony przez lipofilność substancji dyfundującej (tj. współczynnik dystrybucji olej/woda) i gradient stężenia, może być ograniczony bardzo niską rozpuszczalnością substancji w wodzie (co zapobiega przenikaniu leku do fazy wodnej membrany)
3. Związki niepolarne łatwo dyfundują, jony są trudne do dyfuzji.
Każda dyfuzja (zarówno wody, jak i lipidów) podlega prawu dyfuzji Ficka:
Szybkość dyfuzji - liczba cząsteczek leku przenoszonych w jednostce czasu; С 1 - stężenie substancji na zewnątrz membrany, С 2 - stężenie substancji wewnątrz membrany.
Wniosek z prawa Ficka:
1) filtracja leku jest tym większa, im większe jest jego stężenie w miejscu wstrzyknięcia (S powierzchni wchłoniętej w jelicie jest większe niż w żołądku, dlatego wchłanianie leku do jelita jest szybsze)
2) im wyższe stężenie leku w miejscu wstrzyknięcia, tym wyższa filtracja leku
3) filtracja leków jest tym większa, im mniejsza jest grubość błony biologicznej do pokonania (grubość bariery w pęcherzykach płucnych jest znacznie mniejsza niż w skórze, dlatego szybkość wchłaniania jest wyższa w płuca)
Transport aktywny- transfer leku niezależnie od gradientu stężeń za pomocą energia ATP, jest charakterystyczna dla hydrofilowych cząsteczek polarnych, szeregu jonów nieorganicznych, cukrów, aminokwasów, pirymidyn.
Cechuje: a) selektywność dla niektórych związków b) możliwość konkurowania dwóch substancji o jeden mechanizm transportu c) nasycenie przy wysokich stężeniach substancji d) możliwość transportu wbrew gradientowi stężeń e) zużycie energii.
7. Centralnym postulatem farmakokinetyki jest stężenie leku we krwi - główny parametr kontroli efektu terapeutycznego. Problemy rozwiązywane na podstawie znajomości tego postulatu.
Centralny postulat (dogmat) farmakokinetyki: stężenie leku w osoczu krwi określa (ilościowo) działanie farmakologiczne.
W większości przypadków szybkość wchłaniania, dystrybucji, metabolizmu i wydalania leków jest proporcjonalna do ich stężenia w osoczu krwi (przestrzega prawa działania masy), więc wiedząc, że jest to możliwe:
1) określić okres półtrwania (dla leków o kinetyce pierwszego rzędu)
2) wyjaśnić czas trwania niektórych toksycznych efektów leków (dla leków w dużych dawkach o kinetyce nasycenia)
[C] plazma → [C] na cel → efekt
Określony prawami jest określony przez z-us farmakodynamiki
Dystrybucja
Cele: nauczyć się kontrolować stężenie leku we krwi.
Modele farmakokinetyczne (jednokomorowe i dwukomorowe), ilościowe prawa wchłaniania i eliminacji leków.
Cały organizm jest jednym, jednorodnym pojemnikiem. Założenia: 1) następuje szybki dynamiczny rozwój między zawartością leku w ... 2) lek jest szybko i równomiernie rozprowadzany w całej objętości krwiDystrybucja leków w organizmie. Przedziały, ligandy. Główne determinanty dystrybucji.
Przedziały dystrybucyjne: 1. Przestrzeń zewnątrzkomórkowa (osocze, płyn międzykomórkowy) 2. Komórki (cytoplazma, błona organelli)Stała eliminacji, jej istota, wymiar, związek z innymi parametrami farmakokinetycznymi.
Stała szybkości eliminacji(kel, min -1) - pokazuje, jaka część leków jest wydalana z organizmu w jednostce czasu Þ Kel = A out / A total, gdzie A out to ilość uwalnianych leków w jednostkach. czas, a całkowita - całkowita ilość leków w organizmie.
Wartość k el zwykle określa się rozwiązując równanie farmakokinetyczne opisujące proces eliminacji leku z krwi, dlatego k el nazywa się modelowym indeksem kinetycznym. Kel nie jest bezpośrednio związany z planowaniem schematu dawkowania, ale jego wartość jest wykorzystywana do obliczania innych parametrów farmakokinetycznych.
Stała eliminacji jest wprost proporcjonalna do klirensu i odwrotnie proporcjonalna do objętości dystrybucji (z definicji klirensu): Kel = CL / Vd; = godzina -1 / min -1 = ułamek na godzinę.
Okres półtrwania leków, jego istota, wymiar, związek z innymi parametrami farmakokinetycznymi.
Okres półeliminacji(t ½, min) to czas potrzebny do zmniejszenia stężenia leków we krwi dokładnie o połowę. W tym przypadku nie ma znaczenia, w jaki sposób osiąga się zmniejszenie stężenia - za pomocą biotransformacji, wydalania lub połączenia obu procesów.
Okres półtrwania określa wzór:
![]()
Okres półtrwania jest najważniejszym parametrem farmakokinetycznym, który umożliwia:
b) określić czas całkowitego wyeliminowania leku
c) przewidzieć stężenie leków w dowolnym momencie (dla leków o kinetyce pierwszego rzędu)
Klirens jako główny parametr farmakokinetyczny do zarządzania schematem dawkowania. Jego istota, wymiar i związek z innymi parametrami farmakokinetycznymi.
Luz(Cl, ml / min) - objętość krwi, która jest usuwana z leków na jednostkę czasu.
Bo osocze (krew) jest „widoczną” częścią objętości dystrybucji, następnie klirens to ułamek objętości dystrybucji, z którego lek jest uwalniany w jednostce czasu. Jeśli oznaczymy całkowitą ilość leku w organizmie przez I ogólnie, a kwota, która została przydzielona po I vyd, następnie:
Z drugiej strony z definicji objętości dystrybucji wynika, że całkowita ilość leku w organizmie wynosi Suma = V d ´C ter / plazma... Podstawiając tę wartość do wzoru na klirens, otrzymujemy:
![]() .
.
Zatem klirens jest stosunkiem szybkości eliminacji leku do jego stężenia w osoczu krwi.
W tej formie formuła klirensu służy do obliczenia dawki podtrzymującej leku ( Dp), czyli dawka leku, która powinna zrekompensować utratę leku i utrzymać jego poziom na stałym poziomie:
Szybkość podawania = szybkość eliminacji = Cl'C ter (dawka / min)
D p = szybkość infuzji 't (t jest odstępem między przyjmowaniem leku)
Prześwit jest addytywny, tj. eliminacja substancji z organizmu może nastąpić przy udziale procesów w nerkach, płucach, wątrobie i innych narządach: Cl systemowy = Cl nerkowy. + Cl wątroba + Cl inne.
Odprawa ograniczona z okresem półtrwania leku i objętością dystrybucji: t 1/2 = 0,7 * Vd / Cl.
Dawka. Rodzaje dawek. Jednostki dawkowania leków. Cele dawkowania leków, metody i opcje podawania, odstępy między podawaniem.
Wpływ leków na organizm w dużej mierze zależy od ich dawki.
Dawka- ilość substancji wprowadzonej do organizmu jednorazowo; wyrażone w jednostkach masy, objętości lub konwencjonalnych (biologicznych).
Rodzaje dawek:
a) pojedyncza dawka - ilość substancji na dawkę
b) dawka dzienna - ilość leku przepisana na dzień w jednej lub kilku dawkach
c) dawka oczywiście - całkowita ilość leku na przebieg leczenia
d) dawki terapeutyczne - dawki, w których lek jest stosowany z terapeutycznym lub cele zapobiegawcze(próg lub minimalna skuteczna, średnia terapeutyczna i najwyższa terapeutyczna dawka).
e) dawki toksyczne i śmiertelne - dawki leków, przy których zaczynają wykazywać wyraźne działanie toksyczne lub powodują śmierć organizmu.
f) dawka nasycająca (wstępna) - liczba wstrzykniętych leków, która wypełnia całą objętość dystrybucji organizmu w stężeniu skutecznym (leczniczym): VD = (Css * Vd) / F
g) dawka podtrzymująca – systematycznie podawana ilość leków kompensująca utratę leków klirensem: PD = (Css * Cl * DT) / F
Farmaceutyczne jednostki dawkowania:
1) w gramach lub ułamkach grama narkotyków
2) liczba leków na 1 Kg masa ciała (na przykład 1 mg/kg) lub na jednostkę powierzchni ciała (na przykład 1 mg/m2)
Cele dawkowania leków:
1) określić ilość leków potrzebnych do wywołania pożądanego efekt terapeutyczny z określonym czasem trwania
2) unikać zjawisk zatrucia i skutków ubocznych przy wprowadzaniu leków
Metody podawania leku: 1) dojelitowo 2) pozajelitowo (patrz punkt 5)
Opcje podawania leków:
a) ciągły (przez długotrwałą donaczyniową infuzję leków w kroplówce lub przez automatyczne dozowniki). Przy ciągłym podawaniu leków jego stężenie w organizmie zmienia się płynnie i nie ulega znacznym wahaniom.
b) podawanie przerywane (metodą iniekcji lub bez iniekcji) - podawanie leku w regularnych odstępach czasu (odstępy dawkowania). Przy okresowym podawaniu leków jego stężenie w organizmie stale się zmienia. Po przyjęciu określonej dawki najpierw wzrasta, a następnie stopniowo spada, osiągając wartości minimalne przed kolejnym podaniem leku. Wahania stężenia są tym większe, im większa jest podana dawka leku i odstęp między iniekcjami.
Interwał wprowadzenia- odstęp pomiędzy podawanymi dawkami, zapewniający utrzymanie terapeutycznego stężenia substancji we krwi.
15. Podawanie leków w stałym tempie. Kinetyka stężenia leku we krwi. Stacjonarne stężenie leku we krwi (C ss), czas dotarcia do niego, jego obliczenie i zarządzanie.
 Osobliwością wprowadzania leków w stałym tempie jest płynna zmiana ich stężenia we krwi po podaniu, przy czym:
Osobliwością wprowadzania leków w stałym tempie jest płynna zmiana ich stężenia we krwi po podaniu, przy czym:
1) czas do osiągnięcia stężenia leku w stanie stacjonarnym wynosi 4-5t ½ i nie zależy od szybkości wlewu (wielkości podanej dawki)
2) wraz ze wzrostem szybkości wlewu (dawki wstrzykniętej) wartość C SS również wzrasta proporcjonalną liczbę razy
3) eliminacja leku z organizmu po zakończeniu wlewu trwa 4-5t ½.
Сss - równowaga stacjonarna koncentracja- stężenie leków osiągane przy szybkości podawania równej szybkości wydalania, a więc:
![]() (z definicji odprawy)
(z definicji odprawy)
Dla każdego kolejnego okresu półtrwania stężenie leku wzrasta o połowę pozostałego stężenia. Wszystkie leki, które są zgodne z prawem eliminacji pierwszego rzędu, są osiągnie Css w 4-5 okresach półtrwania.
Podejścia do zarządzania poziomem Css: zmień podawaną dawkę leków lub przerwę w podawaniu
16. Przerywane podawanie leków. Kinetyka stężenia leków we krwi, zakres stężeń terapeutycznych i toksycznych. Obliczanie stężenia stacjonarnego (C ss), granic jego wahań i jego kontrola. Odpowiednia dyskretna przerwa w dozowaniu.
 Wahania stężenia leków w osoczu krwi: 1 - ze stałą kroplówką dożylną; 2 - z ułamkowym wprowadzeniem tej samej dziennej dawki w odstępie 8 godzin, 3 - z wprowadzeniem dziennej dawki w odstępie 24 godzin.
Wahania stężenia leków w osoczu krwi: 1 - ze stałą kroplówką dożylną; 2 - z ułamkowym wprowadzeniem tej samej dziennej dawki w odstępie 8 godzin, 3 - z wprowadzeniem dziennej dawki w odstępie 24 godzin.
Okresowe podawanie leku- wprowadzenie pewna ilość Narkotyki w pewnych odstępach czasu.
Równowagowe stężenie w stanie stacjonarnym osiąga się po 4-5 okresach połowicznej eliminacji, czas do jego osiągnięcia nie zależy od dawki (na początku, gdy poziom stężenia leku jest niski, szybkość jego eliminacji jest również niska; jak zwiększa się ilość substancji w organizmie, wzrasta również tempo jej eliminacji, dlatego wcześnie lub późno przyjdzie moment, w którym zwiększone tempo eliminacji zrównoważy podaną dawkę leku i dalszy wzrost stężenia zatrzyma się)
Css jest wprost proporcjonalna do dawki leku i odwrotnie proporcjonalna do przerwy w podawaniu i klirensu leku.
Granice huśtawki CSS: ![]() ; C ss min = C ss max × (1 - e-mail). Wahania stężenia leku są proporcjonalne do T / t 1/2.
; C ss min = C ss max × (1 - e-mail). Wahania stężenia leku są proporcjonalne do T / t 1/2.
Zasięg terapeutyczny (korytarz bezpieczeństwa, okno terapeutyczne)- Jest to zakres stężeń od minimalnego terapeutycznego do wywołującego pierwsze oznaki skutków ubocznych.
Zasięg toksyczny- zakres stężeń od najwyższych terapeutycznych do śmiertelnych.
Odpowiednie podawanie dyskretnych dawek: sposób podawania, w którym wahania stężenia leku we krwi mieszczą się w zakresie terapeutycznym. Aby określić odpowiedni schemat podawania leku, konieczne jest obliczenie. Różnica między Css max i Css min nie powinna przekraczać 2Css.
Kontrolowanie fluktuacji CSS:
Zakres wahań Css jest wprost proporcjonalny do dawki leku i odwrotnie proporcjonalny do odstępu jego podawania.
1. Zmień dawkę leków: wraz ze wzrostem dawki leku zakres wahań jego Css proporcjonalnie wzrasta
2. Zmień interwał podawania leku: wraz ze wzrostem przerwy w podawaniu leku zakres wahań jego Css proporcjonalnie maleje
Jednoczesna zmiana dawki i przerwy w podawaniu
Dawka początkowa (ładująca). Znaczenie terapeutyczne, obliczanie parametrów farmakokinetycznych, warunki i ograniczenia jego stosowania.
Dawka początkowa (ładująca)- dawka podana jednorazowo i wypełnia całą objętość dystrybucji w aktualnym stężeniu terapeutycznym. VD = (Css * Vd) / F; = mg / l, = l / kg
Znaczenie terapeutyczne: dawka początkowa szybko zapewnia skuteczne terapeutyczne stężenie leków we krwi, co umożliwia np. szybkie powstrzymanie ataku astmy, arytmii itp.
Dawkę początkową można podać jednorazowo tylko wtedy, gdy proces dystrybucji substancji jest ignorowany
Ograniczenie korzystania z VD: jeśli lek jest dystrybuowany znacznie wolniej niż jego wejście do krwioobiegu wprowadzenie od razu całej dawki nasycającej (zwłaszcza dożylnie) spowoduje powstanie stężenia znacznie wyższego od terapeutycznego i spowoduje wystąpienie efektów toksycznych. Warunki użytkowania VD: zatem wprowadzenie dawek obciążeniowych zawsze powinien być powolny lub ułamkowy.
Dawki podtrzymujące, ich znaczenie terapeutyczne i obliczanie optymalnego schematu dawkowania.
Sens terapeutyczny: PD kompensuje straty z klirensem w okresie między wstrzyknięciami leku. Obliczanie optymalnej dawki leków (w celu szybkiego złagodzenia ataku): ... 1. Oblicz VD: VD = (Css * Vd) / FRóżnice osobnicze, wiekowe i płciowe w farmakokinetyce leków. Korekty obliczania poszczególnych wartości objętości dystrybucji leków.
2. Różnice płci w działaniu leków. Dla kobiet charakterystyczna jest mniejsza masa ciała niż dla mężczyzn, dlatego wielkość dawek leku dla nich powinna ... 3. Stany patologiczne organizm i działanie leków a) choroba wątroby: leki F z powodu wyłączenia metabolizmu przedukładowego, część niezwiązanych leków z powodu braku ...Klirens nerkowy leków, mechanizmy, ich cechy ilościowe i jakościowe.
Mechanizmy klirensu nerkowego i ich cechy: 1. Filtracja: leki uwalniane tylko przez filtrację (insulina) będą miały klirens, ... Wyznaczone przez: przepływ krwi przez nerki, niezwiązaną frakcję leku i zdolność filtracyjną nerek.Czynniki wpływające na klirens nerkowy leków. Zależność klirensu od właściwości fizykochemicznych leków.
a) filtracja kłębuszkowa b) szybkość przepływu krwi przez nerki c) maksymalna szybkość wydzielaniaKlirens leków wątrobowych, jego uwarunkowania i ograniczenia. Cykl leków jelitowo-wątrobowych.
1) metabolizm (biotransformacja) przez utlenianie, redukcję, alkilację, hydrolizę, koniugację itp. Główna strategia metabolizmu ksenobiotyków: substancje niepolarne ® polar ... 2) wydzielanie (wydalanie nieprzekształconych substancji do żółci)Korekta terapii lekowej w przypadku uszkodzenia wątroby i innych stanów patologicznych.
Aby skorygować schemat dawkowania w przypadku choroby nerek, patrz paragraf 26 powyżej, ogólne zasady korekta - w.25. Korekta schematu dawkowania pod kontrolą całkowitego klirensu leku: Korekta dawki: Dind. = Dtypowe × Clind. / Cltypowe.Korekta schematu dawkowania pod kontrolą resztkowej czynności nerek.
Znamy: a) resztkową czynność nerek, którą określa klirens kreatyniny w danym... b) klirens całkowity danego leku (CLP/całkowity) oraz udział klirensu nerkowego leku w klirensie całkowitymIndywidualna strategia terapii lekowej.
Wyznanie ważna rola koncentracja jako łącze łączące farmakokinetyka i farmakodynamika przyczynia się do stworzenia strategii stężenia docelowego – optymalizacji dawki u danego pacjenta na podstawie pomiaru stężenia leku. Składa się z następujących etapów:
1. Wybór stężenia docelowego
2. Oblicz Vd i Cl na podstawie typowych wartości i dokonaj korekty uwzględniając takie czynniki, jak masa ciała i czynność nerek.
3. Wpisanie dawki nasycającej lub podtrzymującej, obliczonej z uwzględnieniem wartości TC, Vd i Cl.
4. Rejestracja reakcji pacjenta i określenie stężenia leku
5. Weryfikacja Vd i Cl na podstawie wyników pomiarów stężeń.
6. Powtórz kroki 3-6, aby dostosować dawkę podtrzymującą wymaganą dla optymalnej odpowiedzi na lek.
Biotransformacja leków, jej biologiczne znaczenie, główne kierunki i wpływ na działanie leków. Główne fazy przemian metabolicznych leków w organizmie.
Biotransformacja leków – przemiany chemiczne leków w organizmie.
Biologiczne znaczenie biotransformacji leków: tworzenie substratu dogodnego do późniejszej utylizacji (jako materiał energetyczny lub plastik) lub przyspieszenie eliminacji leków z organizmu.
Główny kierunek przemian metabolicznych leków: leki niepolarne → polarne (hydrofilowe) metabolity wydalane z moczem.
Istnieją dwie fazy reakcji metabolicznych leków:
1) przemiany metaboliczne (reakcje niesyntetyczne, faza 1) – przemiany substancji w wyniku mikrosomalnego i pozamikrosomalnego utleniania, redukcji i hydrolizy
2) koniugacja (reakcje syntezy, faza 2) - proces biosyntezy, któremu towarzyszy dodanie do leku lub jego metabolitów szeregu grup chemicznych lub cząsteczek związków endogennych przez a) tworzenie glukuronidów b) estry glicerolu c) sulfoestry d ) acetylacja e) metylacja
Wpływ biotransformacji na aktywność farmakologiczną leków:
1) najczęściej metabolity biotransformacji nie wykazują aktywności farmakologicznej lub ich aktywność jest zmniejszona w porównaniu z substancją wyjściową
2) w niektórych przypadkach metabolity mogą zachować aktywność, a nawet przewyższyć aktywność substancji macierzystej (kodeina jest metabolizowana do bardziej aktywnej farmakologicznie morfiny)
3) czasami podczas biotransformacji powstają substancje toksyczne (metabolity izoniazydu, lidokainy)
4) czasami podczas biotransformacji powstają metabolity o przeciwnych właściwościach farmakologicznych (metabolity nieselektywnych agonistów b 2 - receptory adrenergiczne mają właściwości blokerów tych receptorów)
5) szereg substancji to proleki, które początkowo nie dają efektów farmakologicznych, ale w toku biotransformacji są przekształcane w substancje biologicznie czynne (nieaktywna L-dopa, przenikając przez BBB, zamienia się w mózgu w aktywną dopaminę, natomiast tam nie występują ogólnoustrojowe działania dopaminy).
Kliniczne znaczenie biotransformacji leków. Czynniki wpływające na ich btotransformację. Metaboliczne interakcje leków.
Wpływ na biotransformację leków różne czynniki: a) stan czynnościowy wątroby: w jej chorobach klirens leków jest zwykle… b) wpływ czynników środowiskowych: palenie sprzyja indukcji cytochromu P450, w wyniku czego metabolizm leków w . ...Sposoby i mechanizmy eliminacji leku z organizmu. Możliwości zarządzania eliminacją leków.
Sposoby i mechanizmy wydalania leków: eliminacja leków przez wątrobę i nerki oraz niektóre inne narządy:
a) przez nerki przez filtrację, sekrecję, reabsorpcję
b) przez wątrobę przez biotransformację, wydalanie z żółcią
c) przez płuca, ślinę, pot, mleko itp. przez wydzielanie, parowanie
Możliwości zarządzania procesami odstawienia leków:
1. Kontrola pH: w moczu zasadowym zwiększa się wydalanie związków kwaśnych, w moczu kwaśnym wydalanie związków zasadowych
2.aplikacja leki żółciopędne(cholenzym, allochol)
3. hemodializa, dializa otrzewnowa, hemosorpcja, limfosorpcja
4. wymuszona diureza (IV NaCl lub glukoza do obciążenie wodą+ furosemid lub mannitol)
5. płukanie żołądka, stosowanie lewatyw
Pojęcie receptorów w farmakologii, molekularna natura receptorów, mechanizmy sygnalizacyjne działania leków (rodzaje sygnalizacji transbłonowej i przekaźniki wtórne).
Receptory - składniki molekularne komórki lub organizmu, które oddziałują z lekami i wywołują szereg zdarzeń biochemicznych prowadzących do rozwoju efektu farmakologicznego.
Pojęcie receptorów w farmakologii:
1. Receptory określają ilościowe wzorce działania leku
2. Receptory odpowiadają za selektywność działania leków
3. Receptory pośredniczą w działaniu antagonistów farmakologicznych
Pojęcie receptorów jest podstawą celowego stosowania leków wpływających na procesy regulacyjne, biochemiczne i komunikację.
Molekularna natura receptorów:
1.białka regulatorowe, mediatory działania różnych sygnałów chemicznych: neuroprzekaźniki, hormony, autokoidy
2.enzymy i transbłonowe białka transportujące (Na+, K+ATPaza)
3.białka strukturalne (tubulina, białka cytoszkieletu, powierzchnia komórki)
4.białka jądrowe i kwasy nukleinowe
Mechanizmy sygnalizacyjne działania leku:
1) przenikanie rozpuszczalnych w tłuszczach ligandów przez błonę i ich wpływ na receptory wewnątrzkomórkowe.
2) cząsteczka sygnalizacyjna wiąże się z domeną zewnątrzkomórkową białka transbłonowego i aktywuje aktywność enzymatyczną jego domeny cytoplazmatycznej.
3) cząsteczka sygnalizacyjna wiąże się z kanałem jonowym i reguluje jego otwarcie.
4) cząsteczka sygnalizacyjna wiąże się z receptorem na powierzchni komórki, który jest sprzężony z enzymem efektorowym przez białko G. Białko G aktywuje wtórnego posłańca.
Rodzaje sygnalizacji transbłonowej:
a) przez receptory 1-TMS z aktywnością kinazy tyrozynowej i bez niej
b) przez receptory 7-TMS związane z białkiem G
c) przez kanały jonowe (zależne od liganda, zależne od napięcia, styki szczelinowe)
Pośrednicy wtórni: cAMP, jony Ca2+, DAG, IF3.
Fizykochemiczne i chemiczne mechanizmy działania substancji leczniczych.
Główne efekty farmakologiczne: 1) narkotyczne 2) ogólne depresyjne 3) paraliżujące 4) miejscowo drażniące 5) działanie błoniaste. Charakter chemiczny substancji: chemicznie obojętne węglowodory, etery, alkohole, ... Mechanizm działania - odwracalne zniszczenie błon.Selektywność i specyficzność działania leków. Terapeutyczne, uboczne i toksyczne działanie leków, ich istota z punktu widzenia koncepcji receptorów. Strategia terapeutyczna zwalczania skutków ubocznych i toksycznych leków.
Specyficzność–Wiązanie leków ze ściśle określonym typem receptora.
Selektywność- potrafi wiązać leki z jednym lub kilkoma rodzajami receptorów dokładniej niż inne.
Lepiej jest używać terminu selektywność, ponieważ jest mało prawdopodobne, aby jakakolwiek cząsteczka leku mogła wiązać się tylko z jednym typem cząsteczki receptora, ponieważ liczba potencjalnych receptorów u każdego pacjenta jest astronomiczna.
Działanie terapeutyczne- główny pożądany efekt farmakologiczny oczekiwany od danego preparatu farmakologicznego.
Skutki uboczne- te efekty, które występują, gdy substancje są stosowane w dawkach terapeutycznych i stanowią spektrum ich działania farmakologicznego.
Efekty toksyczne- działania niepożądane objawiające się tym lekiem, gdy opuszcza on zakres terapeutyczny.
Zależności między działaniem terapeutycznym a toksycznym leków na podstawie analizy mechanizmów receptorowo-efektorowych:
1) działanie terapeutyczne i toksyczne, w którym pośredniczy ten sam mechanizm receptorowo-efektorowy (prazosyna działa jako alfa-selektywny antagonista na naczyniowe receptory SMC i ma działanie hipotensyjne w nadciśnieniu pierwotnym, ale przy dużych dawkach pacjent może doświadczać niedociśnienia ortostatycznego)
2) działanie terapeutyczne i toksyczne pośredniczone przez identyczne receptory, ale różne tkaniny lub różnymi szlakami efektorowymi (glikozydy nasercowe są stosowane w celu zwiększenia kurczliwości mięśnia sercowego, jednocześnie zaburzają funkcję przewodu pokarmowego, widzenie z powodu blokady Na+/K+-ATPazy błony komórkowej)
3) działanie terapeutyczne i toksyczne, pośredniczone różne rodzaje receptory (na przykład noradrenalina ma działanie nadciśnieniowe przez 1-Ar, ale jednocześnie powoduje tachykardię przez b 1-Ar)
Strategia terapeutyczna w zwalczaniu terapeutycznych i skutki uboczne LS:
1. Lek należy zawsze podawać w najmniejszej dawce, która powoduje akceptowalny efekt terapeutyczny.
2. Zmniejszenie dawki jednego leku z powodu wyznaczenia innego leku z podobne działanie, ale przez inne receptory io innym profilu toksyczności.
3. Selektywność działania leku można zwiększyć kontrolując stężenie leku w rejonie receptorów różnych części ciała ( aplikacja lokalna LS - wziewne stosowanie salbutamolu na astmę oskrzelową)
32. Terminy i pojęcia farmakologii ilościowej: efekt, skuteczność, aktywność, agonista (pełny, częściowy), antagonista. Kliniczna różnica między pojęciami działania i skuteczności leków.
 Efekt (odpowiedź)- ilościowa wydajność reakcji interakcji komórki, narządu, układu lub organizmu ze środkiem farmakologicznym.
Efekt (odpowiedź)- ilościowa wydajność reakcji interakcji komórki, narządu, układu lub organizmu ze środkiem farmakologicznym.
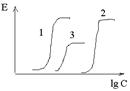
Efektywność- miara reakcji wzdłuż osi efektu - wielkość odpowiedzi układu biologicznego na efekt farmakologiczny; jest to zdolność leku do zapewnienia maksymalnego możliwego efektu... Te. w rzeczywistości jest to maksymalna wielkość efektu, jaką można osiągnąć przy podawaniu danego leku. Numerycznie scharakteryzowany wartością E max. Im wyższe E max, tym wyższa skuteczność leku.
Działalność- miara wrażliwości na leki wzdłuż osi stężenia, charakteryzuje powinowactwo (powinowactwo liganda do receptora), pokazuje, jaka dawka (stężenie) leku jest zdolna do wywołania efektu standardowego równego 50% maksymalna możliwa dla tego leku. Numerycznie scharakteryzowane wartością EC 50 lub ED 50. Im wyższa aktywność leku, tym mniejsza jego dawka jest wymagana do odtworzenia efektu terapeutycznego.
Wydajność: 1 = 2> 3
Aktywność: 1> 3> 2
W praktyce klinicznej ważniejsza jest znajomość skuteczności niż działania, ponieważ bardziej interesuje nas zdolność leków do wywoływania określonego efektu w organizmie.
Agonista- ligand, który wiąże się z receptorem i wywołuje odpowiedź biologiczną, aktywację układu fizjologicznego. Pełen agonista- maksymalna odpowiedź, częściowy- powodują mniejszą reakcję, nawet gdy wszystkie receptory są zajęte.
 Antagonista- ligandy zajmujące receptory lub zmieniające je w taki sposób, że tracą zdolność oddziaływania z innymi ligandami, ale same nie wywołują reakcji biologicznej (blokują działanie agonistów).
Antagonista- ligandy zajmujące receptory lub zmieniające je w taki sposób, że tracą zdolność oddziaływania z innymi ligandami, ale same nie wywołują reakcji biologicznej (blokują działanie agonistów).
Konkurencyjni antagoniści- oddziałują odwracalnie z receptorami i w ten sposób konkurują z agonistami. Zwiększenie stężenia agonisty może całkowicie wyeliminować działanie antagonisty. Konkurencyjny antagonista przesuwa krzywą dawka-efekt dla agonisty, zwiększa EC50, nie wpływa na Emax.
Niekonkurencyjni antagoniści- nieodwracalnie zmieniają powinowactwo receptorów do agonisty, często nie dochodzi do wiązania z miejscem aktywnym receptora, wzrost stężenia agonisty nie eliminuje działania antagonisty. Niekompetycyjny antagonista zmniejsza Emax, nie zmienia EC50, a krzywa dawka-odpowiedź jest skompresowana wokół osi pionowej.
33. Ilościowe wzorce działania leków. Prawo zmniejszania odpowiedzi systemów biologicznych. Model Clarka i jego konsekwencje. Ogólna forma koncentracja zależności - efekt we współrzędnych normalnych i lognormalnych.
Model Clarka-Ariensa:
1. Oddziaływanie między ligandem (L) a receptorem (R) jest odwracalne.
2. Wszystkie receptory dla danego ligandu są równoważne i niezależne (ich nasycenie nie wpływa na inne receptory).
3. Efekt jest wprost proporcjonalny do liczby zajętych receptorów.
4. Ligand występuje w dwóch stanach: wolnym i związanym z receptorem.
A), gdzie Kd jest stałą równowagi, Ke jest aktywnością wewnętrzną.
B) Ponieważ wraz ze wzrostem liczby ligandów w pewnym momencie wszystkie receptory będą zajęte, wówczas maksymalna możliwa liczba utworzonych kompleksów ligand-receptor jest opisana wzorem:
= [R] × (1)
Efekt zależy od prawdopodobieństwa aktywacji receptora po związaniu z ligandem, tj. jego aktywność wewnętrzna (Ke), zatem E = Ke ×. W tym przypadku efekt jest maksymalny przy Ke = 1 i minimalny i Ke = 0. To naturalne, że maksymalny efekt jest opisana zależnością Emax = Ke ×, gdzie jest całkowitą liczbą receptorów dla danego ligandu
Efekt zależy również od stężenia ligandu na receptorach [C], dlatego
Z powyższych relacji wynika, że EC 50 = Kd
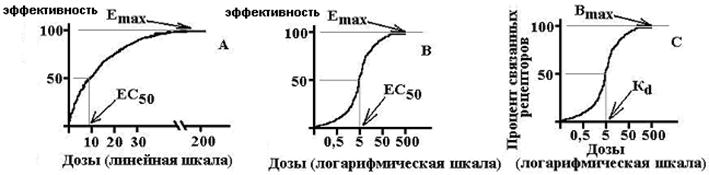
E max to maksymalny efekt, B max to maksymalna liczba związanych receptorów, EC 50 to stężenie leku, przy którym występuje efekt równy połowie maksimum, K d to stała dysocjacji substancji od receptora, przy które 50% receptorów jest związanych.
Prawo malejącej odpowiedzi odpowiada paraboliczna zależność „stężenie - wydajność”. Reakcja na niskie dawki leków zwykle wzrasta wprost proporcjonalnie do dawki... Jednak wraz ze wzrostem dawki wzrost odpowiedzi maleje i ostatecznie można osiągnąć dawkę, przy której nie ma dalszego wzrostu odpowiedzi (ze względu na zajęcie wszystkich receptorów dla danego liganda).
Stopniowa i kwantowa ocena efektu, istoty i zastosowań klinicznych. Kwantyfikacja aktywności i skuteczności leków w praktyce eksperymentalnej i klinicznej.
Wszystkie efekty farmakologiczne można z grubsza podzielić na dwie kategorie:
ale) efekty stopniowe (ciągłe, całkowe)- takie działanie leków, które można zmierzyć ilościowo (działanie leków hipotensyjnych - przez poziom ciśnienia krwi). Opisano stopniową „krzywą dawka-efekt” (patrz str. 33), na podstawie której można ocenić: 1) indywidualną wrażliwość na leki; 2) aktywność narkotykowa; 3) maksymalna skuteczność leku.
b) efekty kwantowe- takie efekty leków, które są dyskretną ilością, cechą jakościową, tj. opisane tylko przez kilka opcji stanów (ból głowy po zażyciu środka przeciwbólowego, albo występuje, albo nie). Opisano krzywą kwantową dawka-efekt, w której odnotowuje się zależność manifestacji efektu w populacji od wartości przyjętej dawki leku. Wykres dawka-efekt ma kształt kopuły i jest identyczny z krzywą rozkładu normalnego Gaussa. Na podstawie krzywej kwantowej można: 1) oszacować populacyjną wrażliwość leków; 2) odnotować obecność efektu przy danej dawce; 3) wybrać średnią dawkę terapeutyczną.
Różnice między stopniową a kwantową charakterystyką efektu dawki:
Ilościową ocenę aktywności i skuteczności leków przeprowadza się na podstawie konstrukcji krzywych dawka-skutek i ich późniejszej oceny (patrz Rozdział 32)
Rodzaje działania leków. Zmiany w działaniu leków, gdy się powtarzają.
1. Przez lokalizację: · lokalnie - efekt, który występuje w miejscu podania leku (zwykle na skórę i ... · resorpcyjny - jest to efekt, który ma lek po wchłonięciu do krwioobiegu lub bezpośrednim wstrzyknięciu do .. .Zależność działania leków od wieku, płci i indywidualnych możliwości organizmu. Znaczenie rytmów dobowych.
Dzieci - dziedzina farmakologii, która bada charakterystykę działania leków na organizm dziecka, nazywana jest farmakologią pediatryczną ... · osoby starsze - u osób starszych i podeszły wiek farmakokinetyka ... 2. Płeć - eksperymenty na zwierzętach i obserwacje kliniczne pokazują, że istnieją różnice między płciami w ...Zmienność i zmienność działania leków. Hipo- i nadreaktywność, tolerancja i tachyfilaksja, nadwrażliwość i idiosynkrazja. Przyczyny zmienności działania leków i racjonalnej strategii terapii.
Zmienność działania leku – odzwierciedla różnicę w działaniu farmakologicznym jednego leku u różnych osób.
Zmienność działania leku – odzwierciedla zdolność leku do wywoływania efektu odbiegającego od jego typowych efektów farmakologicznych.
Warianty zmienności i zmienności działania leku:
1. Hiporeaktywność- zmniejszenie działania danej dawki leków w porównaniu z efektem obserwowanym u większości pacjentów.
2. Nadreaktywność- wzrost działania danej dawki leków w porównaniu z efektem obserwowanym u większości pacjentów.
3. Tolerancja- zmniejszenie reakcji organizmu na powtarzające się zastrzyki leków; aby przywrócić odpowiedź na leki, musi być podawany w coraz większych dawkach.
4. Tachyfilaksja- stan, w którym częste podawanie leków powoduje rozwój tolerancji po kilku godzinach, ale przy wystarczająco rzadkim podawaniu leków jego działanie jest w pełni zachowane. Rozwój tachyfilaksji jest zwykle związany z wyczerpywaniem się układów efektorowych.
5. Nadwrażliwość- rozwój reakcji alergicznej lub innej reakcji immunologicznej na wielokrotne podawanie leków.
6. Idiosynkrazja- przewrotna reakcja organizmu na leki związane z genetyczną charakterystyką metabolizmu leków lub indywidualną reaktywnością immunologiczną.
Główne przyczyny zmienności działania leku:
1) zmiana stężenia substancji w strefie receptorowej - ze względu na różnice w szybkości wchłaniania, jej dystrybucji, metabolizmie, eliminacji
2) wahania stężenia endogennego ligandu – receptora – propranololu (β-blokera) spowalniającego akcję serca u osób z podwyższony poziom katecholaminy we krwi, ale nie wpływają na tętno w tle u sportowców.
3) zmiany gęstości lub funkcji receptorów.
4) zmiany w składnikach reakcji zlokalizowanych dystalnie do receptora.
Strategia racjonalnej terapii: polega na tym, że w celu wyznaczenia leku i aby przyniósł oczekiwany efekt, należy wziąć pod uwagę indywidualne czynniki organizmu (wiek, płeć itp.), codzienne rytmy osoby, obecność chorób przewlekłych i innych nieprawidłowości. Niezbędne jest również przewidywanie rozwoju efektów nietypowych, biorąc pod uwagę wszystkie możliwe opcje zmienność działania leku.
Ocena bezpieczeństwa leków. Indeks terapeutyczny i standardowe marginesy bezpieczeństwa.
Na stworzenie i wdrożenie nowego leku przeznaczane są ogromne środki – od 100 do 350 milionów dolarów i więcej. Koszty te obejmują robociznę wydatkowaną na ... Aby przejść wszystkie etapy oceny leku muszą odpowiadać główne ... Ocena bezpieczeństwa nowych leków przeprowadzana jest w 2 etapach:Interakcje farmakokinetyczne leków (przykłady).
Interakcja farmakokinetyczna leków jest rodzajem interakcji farmakologicznej, tj. interakcja leków, która objawia się tylko wtedy, gdy razem wchodzą do ludzkiego ciała.
Interakcja farmakokinetyczna zachodzi na etapie wchłaniania, dystrybucji i osadzania, metabolizmu i wydalania.
1. Podczas fazy ssania- ten rodzaj interakcji może prowadzić do zwiększenia lub zmniejszenia ich wchłaniania. Można tego uniknąć, jeśli przerwa między lekami jest przyjmowana co najmniej 4 godziny.
Gdy lek jest podawany doustnie, jego wchłanianie określa:
· pH podłoża- leki niezjonizowane są lepiej wchłaniane w przewodzie pokarmowym niż te zjonizowane, dlatego wzrost pH soku żołądkowego zwiększa wchłanianie słabych zasad i zmniejsza wchłanianie słabych kwasów. Przykład: leki zobojętniające sok żołądkowy, blokery receptorów H2-histaminowych hamują wchłanianie ketokonazolu i innych leków przeciwgrzybiczych, pośrednie antykoagulanty, kwas acetylosalicylowy, barbiturany (prawie całkowicie zapobiegają ich działaniu nasennemu); wzrost pH pożywki poprawia wchłanianie glibutydu, przyspiesza rozpuszczanie otoczki substancji rozpuszczalnych w jelitach.
· bezpośrednia interakcja w przewodzie pokarmowym- tworzenie kompleksów chelatowych i związków, które nie są wchłaniane w przewodzie pokarmowym. NS Przykłady: węgiel aktywny tworzy nierozpuszczalne związki z lekami, zapobiegając ich wchłanianiu w przypadku zatrucia; tetracykliny oddziałują z wapniem, glinem, żelazem, magnezem tworząc kompleksy chelatowe, dlatego ich wchłanianie zmniejsza się przy stosowaniu środków zobojętniających kwasy, preparatów bizmutu; fluorochinolony + leki zobojętniające lub sukralfat = zmniejszona skuteczność antybiotykoterapii.
· motoryka przewodu pokarmowego- może powodować przyspieszenie lub spowolnienie wchłaniania leku. Przykłady: prokinetyka (metoklopromid) przyspiesza wchłanianie szybko wchłanianych leków (etanol, paracetamol, tetracyklina) i spowalnia te wolno wchłaniane (digoksyna, cymetydyna); środki przeczyszczające zmniejszają wchłanianie i biodostępność leków; podczas przyjmowania leków antycholinergicznych blokery receptorów histaminowych H 2 (wydłużenie czasu przejścia leków przez przewód pokarmowy) zwiększają biodostępność i wchłanianie glikozydów nasercowych, preparatów żelaza, co może prowadzić do manifestacji działania toksycznego.
· mikroflora jelitowa - jest bezpośrednio zaangażowana w wchłanianie leków, dlatego każda dysbioza objawia się zaburzeniami wchłaniania leków. Przykłady: digoksyna + erytromycyna = wzrost stężenia digoksyny we krwi i rozwój działań niepożądanych; doustne środki antykoncepcyjne + szerokie spektrum AB = zmniejszony efekt antykoncepcyjny
· uszkodzenie jelitowego CO- hamuje wchłanianie niektórych leków. Przykłady: cytostatyki (cyklofosfamid) hamują wchłanianie digoksyny; upośledzone wchłanianie preparatów żelaza, cyjanokobalaminy, kwasu foliowego.
Na etapie dystrybucji i depozytu
· Konkurencyjne przemieszczenie z wiązania z albuminą osocza krwi – jeśli lek jest związany z białkami poniżej 90%, to przemieszczenie z wiązania z nim nie doprowadzi do… · przemieszczenie z wiązania z białkami w tkankach: chinidyna wypiera digoksyna + ... 3. Na etapie metabolizmu - leki mogą zwiększać lub zmniejszać aktywność układy enzymatyczne bierze udział w metabolizmie leków (...Na etapie wylęgu
· Zmiany wydzielania kanalikowego - chinidyna + digoksyna = zwiększone stężenie digoksyny we krwi i rozwój efektów toksycznych (chinidyna... · zmiany reabsorpcji kanalikowej - tylko reabsorpcja to... 40. Farmakodynamiczne oddziaływanie leków Antagonizm, synergizm , ich rodzaje. Charakter zmiany ...Skutki uboczne i toksyczne substancji leczniczych. Teratogenne, embriotoksyczne, mutagenne działanie leków. Medyczne i aspekty społeczne walka z uzależnieniem od narkotyków, narkomanii i alkoholizmem. Pojęcie nadużywania substancji.
Skutki uboczne- te efekty, które występują, gdy substancje są stosowane w dawkach terapeutycznych i stanowią spektrum ich działania farmakologicznego, mogą być pierwotne i wtórne:
a) pierwotne skutki uboczne – jako bezpośrednia konsekwencja oddziaływania tego leku na określonym podłożu (hiposaliwacja przy użyciu atropiny w celu wyeliminowania bradyarytmii)
b) wtórne skutki uboczne – pośrednio występujące działania niepożądane (AB, tłumienie prawidłowej mikroflory, może prowadzić do nadkażenia)
Efekty toksyczne- działania niepożądane objawiające się tym lekiem, gdy opuszcza on zakres terapeutyczny (przedawkowanie leku)
Selektywność działania leku zależy od jego dawki. Im wyższa dawka leku, tym mniej selektywny staje się.
Działanie teratogenne- zdolność leków podawanych kobiecie w ciąży do wywoływania anatomicznych nieprawidłowości rozwoju płodu (talidomid: fokomelia, leki przeciwblastoma: liczne wady)
Działanie embriotoksyczne- działanie niepożądane niezwiązane z naruszeniem organogenezy w pierwszych trzech miesiącach ciąży. Więcej późniejsze daty manifestuje się działanie fetotoksyczne.
Działanie mutagenne leków- uszkodzenie komórki rozrodczej i jej aparatu genetycznego leków, objawiające się zmianą genotypu potomstwa (adrenalina, cytostatyki).
Działanie rakotwórcze leków- zdolność niektórych leków do indukowania kancerogenezy.
1) Uzależnienie od narkotyków- stan psychiczny i/lub stan fizyczny, który jest konsekwencją wpływu na organizm leków i charakteryzuje się specyficznymi reakcjami behawioralnymi, trudno jest przezwyciężyć chęć ponownego zażywania leków w celu uzyskania specjalnego efektu psychicznego lub uniknięcia dyskomfortu w przypadku braku leków w ciele. Uzależnienie od narkotyków charakteryzuje się:
ale) uzależnienie psychiczne- rozwój niepokoju emocjonalnego po zaprzestaniu zażywania narkotyków. Człowiek czuje się pusty, popada w depresję, odczuwa strach, niepokój, jego zachowanie staje się agresywne. Wszystkie te objawy psychopatologiczne powstają na tle myśli o potrzebie wstrzykiwania sobie narkotyków, które spowodowały uzależnienie. Chęć zażywania narkotyków może wahać się od proste pragnienie do namiętnego pragnienia zażywania narkotyków, które pochłania wszystkie inne potrzeby i zmienia sens życia człowieka. Uważa się, że uzależnienie psychiczne rozwija się, gdy dana osoba uświadamia sobie, że może osiągnąć optymalne samopoczucie wyłącznie poprzez wprowadzenie leków. Fundacja uzależnienie psychiczne- wiara danej osoby w działanie leku (w literaturze opisano przypadki rozwoju uzależnienia psychicznego od placebo).
b) uzależnienie fizyczne- naruszenie normy stan fizjologiczny organizm, który wymaga stałej obecności w nim leków, aby utrzymać stan równowagi fizjologicznej. Odstawienie leku powoduje rozwój specyficznego zespołu objawów - zespołu abstynencyjnego - zespołu zaburzeń psychicznych i neurowegetatywnych w postaci dysfunkcji bocznej, przeciwieństwo tego, co jest charakterystyczne dla działania (morfina likwiduje ból, hamuje ośrodek oddechowy, zwęża źrenice, powoduje zaparcia; z wycofaniem pacjent odczuwa rozdzierający ból, częste głośne oddychanie, rozszerzenie źrenic i uporczywą biegunkę)
w) tolerancja... Tolerancja na leki powodujące uzależnienie od narkotyków jest często przekrojowa, tj. powstaje nie tylko do danego związku chemicznego, ale do wszystkich strukturalnie podobnych związków. Na przykład u pacjentów uzależnionych od morfiny pojawia się tolerancja nie tylko na nią, ale także na inne opioidowe leki przeciwbólowe.
W przypadku rozwoju uzależnienia od narkotyków obecność wszystkich 3 kryteriów nie jest warunek konieczny.
Opioidy, barbiturany, alkohol powodują silne uzależnienie fizyczne i psychiczne oraz tolerancję. Leki przeciwlękowe (diazepam, alprazolam) powodują głównie uzależnienie psychiczne.
2) Uzależnienie (uzależnienie od narkotyków)- Jest to niezwykle ciężka forma uzależnienia, kompulsywne zażywanie narkotyków, charakteryzujące się stale narastającą, nieodpartą chęcią podawania tego leku, zwiększania jego dawki. Kompulsywność pożądania oznacza, że potrzeba podania leku przez pacjenta dominuje nad wszystkimi innymi (nawet żywotnymi) potrzebami. Z punktu widzenia ta definicja, głód morfiny to uzależnienie od narkotyków, podczas gdy głód nikotynowy to uzależnienie od narkotyków.
3) Uzależniony od medycyny- charakteryzuje się mniej intensywnym głodem leków, gdy odmowa przyjmowania leków powoduje jedynie lekkie uczucie dyskomfort, bez rozwoju uzależnienia fizycznego lub szczegółowego obrazu uzależnienia psychicznego. To. uzależnienie obejmuje tę część uzależnienia od narkotyków, która nie pasuje do definicji uzależnienia. Na przykład wspomniane uzależnienie od nikotyny jest formą uzależnienia.
4) Narkomania- nieautoryzowane stosowanie leków w takich dawkach i w sposób, który odbiega od przyjętych standardów medycznych lub społecznych w danej kulturze i podany czas... To. nadużywanie narkotyków obejmuje jedynie społeczne aspekty używania narkotyków. Przykładem nadużycia jest stosowanie sterydów anabolicznych w sporcie lub w celu poprawy sylwetki młodych mężczyzn.
5) Alkoholizm- przewlekłe nadużywanie alkoholu (alkoholu etylowego), prowadzące obecnie do uszkodzenia wielu narządów (wątroby, przewodu pokarmowego, ośrodkowego układu nerwowego, układu krążenia, układ odpornościowy) i towarzyszy temu uzależnienie psychofizyczne.
6) Nadużywanie substancji- przewlekłe nadużywanie różnych narkotyków (m.in. narkotyki, alkohol, halucynogeny), objawiające się różnymi zaburzeniami psychicznymi i somatycznymi, zaburzeniami zachowania, degradacją społeczną.
Leczenie uzależnienia od narkotyków trudne i niewdzięczne zadanie. Nadal nie utworzono skuteczna metodologia, co zapewniłoby powodzenie leczenia u ponad 30-40% pacjentów. Docieram mimo to godne uwagi wyniki jest to możliwe tylko przy pełnej współpracy wysiłków pacjenta, lekarza i tego środowisko socjalne, w którym znajduje się chory (zasada dobrowolności i indywidualności). W sercu nowoczesne techniki kłamstwo następujące zasady:
Metody psychoterapeutyczne i terapii zajęciowej;
Grupowe leczenie i rehabilitacja (anonimowe towarzystwo alkoholików, narkomanów)
Stopniowe lub nagłe odstawienie leku podczas terapii detoksykacyjnej
Terapia substytucyjna (zastępcza narkotyczny powolne i długo działające analogi z ich późniejszym anulowaniem; na przykład tzw. program terapii substytucyjnej metadonem dla uzależnionych od heroiny)
Leczenie specyficznymi antagonistami (nalokson i naltrekson) lub środkami uczulającymi (teturam)
Neurochirurgiczne metody kriodestrukcji zakrętu obręczy i hipokampa
42. Farmaceutyczne interakcje leków. Ostrzeżenia i środki ostrożności dotyczące płynoterapii.
Interakcje farmaceutyczne - rodzaj interakcji związanych z reakcją fizykochemiczną między lekami podczas wytwarzania produktu leczniczego, jeszcze przed wprowadzeniem tych środków do organizmu ludzkiego
ale) typowe błędy prowadzące do niezgodności farmaceutycznej: pisanie skomplikowanych recept, niewłaściwe przechowywanie, nie jest brane pod uwagę możliwość adsorpcji leków na powierzchni plastiku (azotany organiczne)
b) problemy z terapią infuzyjną: mieszanie rozpuszczalnych soli, pochodnych nierozpuszczalnych słabych kwasów lub zasad prowadzi do ich wytrącenia; w płynnych postaciach dawkowania glikozydy nasercowe i alkaloidy ulegają hydrolizie, AB ulega zniszczeniu; pH podłoża (alkaloidy wytrącają się w środowisku alkalicznym)
c) zalecenia: 1) Wszystkie mieszaniny lepiej przygotować ex tempore 2) Najbardziej niezawodnym rozwiązaniem jest jeden lek 3) Przed użyciem należy sprawdzić wszystkie roztwory pod kątem zawiesiny 4) Interakcja może zachodzić bez widocznych zmian w roztworach 5) Leki nie mogą dodać do krwi i roztworów AK 6) W przypadku braku specjalnych instrukcji preparaty należy rozpuścić w 5% roztworze glukozy (pH 3,5-6,5), izotonicznym roztworze NaCl (pH 4,5-7,0).
Roztwór glukozy stabilizowany HCl jest niezgodny z epinefryną, benzylopenicyliną, apomorfiną, kanamycyną, witaminą C, oleandomycyną, glikozydami nasercowymi. Glikozydy nasercowe są niezgodne z atropiną, papaweryną, platifillin. AB są niezgodne z heparyną, hydrokortyzonem. Witaminy z grupy B są ze sobą niekompatybilne, z witaminami PP, C. Witaminy PP i C również są ze sobą niekompatybilne.
Nie można mieszać z innymi lekami: fenotiazydem, chlorpromazyną, barbituranami, preparatami witaminy C, amfoterycyną B, furosemidem, sulfadiazyną, aminofiliną, adrenomimetykami.
Rodzaje farmakoterapii. Deontologiczne problemy farmakoterapii.
1. etiotropowe PT - korekta i eliminacja przyczyny choroby (AB w chorobach zakaźnych) 2. patogenetyczne PT - wpływ na mechanizm rozwoju choroby (inhibitory ...Podstawowe zasady leczenia i profilaktyki zatruć lekami. Terapia antidotum (przykłady).
Klasyfikacja substancji toksycznych (OM):
1. Przynależność do określonych klas związki chemiczne: barbiturany, benzodiazepiny, cyjanki.
2. Według pochodzenia: natura niebiologiczna (kwasy, zasady, sole metali ciężkich), toksyczne produkty odpadowe niektórych MB (toksyna botulinowa), pochodzenie roślinne(alkaloidy, glikozydy), pochodzenia zwierzęcego (jad węża i pszczeli)
3. W zależności od stopnia toksyczności: a) skrajnie toksyczny (DL50< 1 мг/кг) б) высоко токсические (1-50) в) сильно токсические (50-500) г) умеренно токсические (500-5000) д) мало токсические (5000-15000) е) практически нетоксические (> 15.000)
4. Poprzez działanie toksykologiczne: a) paraliżujące nerwy (skurcz oskrzeli, duszenie) b) resorpcyjne skóry c) toksyczne ogólne (konwulsje hipoksyjne, śpiączka, paraliż) d) duszące e) łzowe i drażniące e) psychotropowe (upośledzona aktywność umysłowa, świadomość )
5. W zależności od obszaru preferencyjnego zastosowania: trucizny przemysłowe, pestycydy, trucizny domowe, bojowe środki chemiczne, substancje lecznicze.
6. W zależności od toksyczności leków: Lista A – leki, których przeznaczenie, stosowanie, dawkowanie i przechowywanie, ze względu na ich wysoką toksyczność, należy wykonywać z dużą ostrożnością. Ta sama lista obejmuje leki powodujące uzależnienie od narkotyków; lista B - leki, których mianowanie, stosowanie, dawkowanie i przechowywanie należy przeprowadzać ostrożnie w związku z możliwe komplikacje w przypadku stosowania bez nadzoru lekarskiego.
Selektywnie toksyczne działanie leków.
a) kardiotoksyczne: glikozydy nasercowe, preparaty potasu, leki przeciwdepresyjne
b) neurotoksyczne: środki psychofarmakologiczne, oksychinoliny, aminoglikozydy
c) hepatotoksyczne: tetracykliny, chloramfenikol, erytromycyna, paracetamol
d) nefrotoksyczne: wankomycyna, aminoglikozydy, sulfonamidy
e) gastroenterotoksyczne: steroidowe leki przeciwzapalne, NLPZ, rezerpina
f) hematotoksyczne: cytostatyki, chloramfenikol, sulfonamidy, azotany, azotyny
g) pneumotoksyczne
Toksykokinetyka - bada wchłanianie, dystrybucję, metabolizm i wydalanie leków przyjmowanych w dawkach toksycznych.
Możliwe jest przedostanie się substancji toksycznych do organizmu a) dojelitowo b) pozajelitowo. Szybkość i kompletność wchłaniania odzwierciedla tempo rozwoju efektu toksycznego i jego nasilenie.
Dystrybucja w organizmie: Vd = D / Cmax - rzeczywista objętość, w której trująca substancja jest rozprowadzana w organizmie. Vd> 5-10 l/kg - OM trudno pozwolić na jego usunięcie (leki przeciwdepresyjne, fenotiazyny). Vd< 1 л/кг – ОВ легче удалить из организма (теофиллин, салицилаты, фенобарбитал).
Przedawkować- zmiany w procesach farmakokinetycznych: rozpuszczalność, związek z białkami, metabolizm ® znaczny wzrost wolnej frakcji leków ® działanie toksyczne.
Kinetyka pierwszego rzędu wraz ze wzrostem stężenia leku przekształca się w kinetykę rzędu zerowego.
Etap toksynogenny to terapia detoksykacyjna, etap somatogenny to terapia objawowa.
Toksydynamika . Główne mechanizmy działania toksycznego:
a) mediator: bezpośredni (według rodzaju blokady kompetycyjnej – FOS, psychomimetyki) i pośredni (aktywatory lub inhibitory enzymów)
b) interakcja z biocząsteczkami i strukturami wewnątrzkomórkowymi (substancje hemolityczne)
c) metabolizm według rodzaju letalnej syntezy ( etanol, tiofos)
d) enzymatyczny (jad węża itp.)
Rodzaje działania: lokalne, odruchowe, resorpcyjne.
Klasyfikacja zatruć:
1. Etiopatogenetyczne:
a) przypadkowe (samoleczenie, błędny odbiór)
b) umyślne (w celu popełnienia samobójstwa, zabójstwa, rozwinięcia w ofierze stanu bezradności)
2. Kliniczne:
a) w zależności od szybkości rozwoju zatrucia: ostre (przyjmowanie pojedynczej dawki lub w krótkim odstępie czasu toksycznej dawki substancji), podostre (opóźniony rozwój obraz kliniczny po pojedynczej dawce), przewlekłe
b) w zależności od manifestacji głównego zespołu: uszkodzenie CVS, uszkodzenie DS itp.
c) w zależności od ciężkości stanu pacjenta: łagodny, umiarkowany, ciężki, skrajnie ciężki
3. Nozologiczny: uwzględnia nazwę leku, nazwę grupy substancji
Ogólny mechanizm śmierci w przypadku zatrucia:
a) porażka CVS:
1) obniżenie ciśnienia krwi, hipowolemia naczynia obwodowe, zapaść, brady- lub tachykardia (trójpierścieniowe leki przeciwdepresyjne, beta-blokery, blokery kanału wapniowego)
2) arytmie (częstoskurcz komorowy, migotanie – trójpierścieniowe leki przeciwdepresyjne, teofilina, amfetamina)
b) uszkodzenie ośrodkowego układu nerwowego: otępienie, śpiączka ® depresja oddechowa (leki, barbiturany, alkohol, leki nasenne)
c) drgawki, nadreaktywność i sztywność mięśni ® hipertermia, mioglobinuria, niewydolność nerek, hiperkaliemia
Triada toksykologiczna:
1) czas użytkowania, dawka i historia substancji ®.
2) ocena stanu świadomości według objawów: oddychanie, ciśnienie krwi, temperatura ciała body
3) dane laboratoryjne
Podstawowe zasady leczenia:
I. Pierwszy intensywna opieka : sztuczne oddychanie, masaż serca, terapia przeciwwstrząsowa, kontrola równowagi wodno-elektrolitowej
II. Opóźnione wchłanianie i usuwanie niewchłoniętego OM z organizmu:
Cel: zakończenie kontaktu z OV
1. Droga pozajelitowa:
a) przez płuca:
1) zatrzymaj inhalację
2) substancje drażniące (amoniak, formaldehyd) ® do utrwalania aktywnych ruchów, rozgrzewania, dotleniania i odpieniaczy (w amoniak ocet przeciwpieniący i formaldehyd ma rozcieńczony roztwór amoniaku)
b) przez skórę: zmyć dużą ilością ciepła woda mydłem lub detergentem, swoistymi odtrutkami, neutralizacją i zakończeniem ekspozycji na czynniki na skórze (FOS: przemyty wodą, usunięty 10-15% amoniakiem lub 5-6% roztworem wodorowęglanu sodu wodą; fenolkrezol: olej roślinny lub glikol etylenowy , ale jest to zabronione Olej wazelinowy, KMNO 4: 0,5-1% roztwór kwas askorbinowy lub równe objętości 3% nadtlenku wodoru i 3% roztworu kwas octowy, CCl 4, terpentyna, benzyna: ciepła woda mydlana)
c) w przypadku wstrzyknięcia w kończynę: opaska uciskowa powyżej miejsca wstrzyknięcia
d) w przypadku kontaktu z oczami: płukać ciepłą solanką lub mlekiem przez 10-20 minut, zakroplić środek miejscowo znieczulający; w przypadku kontaktu z kwasami i zasadami nie można ich zneutralizować. Konieczna jest konsultacja z okulistą.
2. Droga dojelitowa: uwolnienie żołądka od OM, przyspieszenie pasażu,
a) usunięcie OM:
1) wstępne pobranie wody. Nie wolno spożywać mleka (z wyjątkiem żrących substancji trujących) i etanolu (z wyjątkiem metanolu).
2) wymioty – wskazane głównie w przypadku zatrucia dużymi tabletkami lub kapsułkami, które nie mogą przejść przez sondę. Może być wywołany odruchem lub wymiotami (NaCl: 1 łyżka stołowa na 1 szklankę wody; syrop Ipecac: dorośli 2 łyżki, dzieci 2 łyżeczki; musztarda: 1-2 łyżeczki na szklankę wody; apomorfina: 5-10 mg/kg podskórnie , z wyjątkiem dzieci poniżej 5 roku życia). Nie wywoływać wymiotów po spożyciu: rozpuszczalniki organiczne - niebezpieczeństwo wdychania, detergenty - substancje pieniące, drgawkowe - niebezpieczeństwo aspiracji, substancje żrące - uszkodzenie przełyku)
3) sonda do płukania żołądka - jest środkiem doraźnym i obowiązkowym. żołądek jest płukany, jeśli od zatrucia minęło nie więcej niż 4-6 godzin, czasem do 10 godzin; w przypadku zatrucia kwas acetylosalicylowy- po 24 godzinach. Pacjent jest wstępnie zaintubowany rurką z nadmuchiwanym mankietem: w śpiączce przy braku kaszlu i odruchu krtaniowego. Żołądek myje się wodą lub roztworem soli o temperaturze 30 ° C, procedura trwa 4 godziny lub dłużej. Pod koniec prania węgiel aktywny i siarczan sodu.
b) zmniejszenie wchłaniania z przewodu pokarmowego: węgiel aktywowany wewnątrz po opróżnieniu żołądka + siarczan sodu lub magnezu. Cechy środków zmniejszających wchłanianie:
1) rozpuszczalniki organiczne: nie wywołują wymiotów, płukanie żołądka po intubacji, węgiel aktywny + parafina ciekła
2) detergenty: nie wywoływać wymiotów i płukania żołądka, należy podać dużo wody + odpieniacze (simetikon)
3) kwasy i zasady: nie wywoływać wymiotów, płukać żołądek przez rurkę, smarować olej roślinny po wprowadzeniu narkotycznego środka przeciwbólowego – jedyne wskazanie do podawania mleka. Do zatrucia kwasami - leki zobojętniające kwasy, do zatrucia zasadami - kwas cytrynowy lub octowy.
III. Usunięcie wchłoniętego OM z organizmu
a) wymuszona diureza (warunki: dostateczny przepływ krwi przez nerki i filtracja kłębuszkowa; wylać 20-25 litrów w ciągu 24 godzin)
b) hemodializa otrzewnowa
c) hemosorpcja
d) wymiana transfuzji krwi
e) wymuszona hiperwentylacja
IV. Terapia objawowa zaburzeń czynnościowych.
2) toksykokinetyczne – przyspieszają biotransformację OM (bromek trimedoksymu, tiosiarczan sodu, etanol, AO) 3) farmakologiczne – atropina, nalokson 4) odtrutki immunologiczneZasady przepisywania leków trujących, odurzających i silnie działających.
Recepty na substancje odurzające (morfina, omnopon, promedol, fenamina, kokaina itp.) i te z nimi równoważne, niezależnie od narkotyków. f- mamy przepisane na specjalne... W przypadku lek. Treść poślubna. alkohol, następnie pieczęć jest kładziona. instytucje. „Dla ... W przypadkach, gdy max. dawka trująca lub silna. przekroczenie dopływu, wymagane. podaj ich liczbę słownie z dodatkiem ...Co zrobimy z otrzymanym materiałem:
Jeśli ten materiał okazał się dla Ciebie przydatny, możesz zapisać go na swojej stronie w sieciach społecznościowych:
Informacje o czasie wchłaniania, dystrybucji i eliminacji, czyli o farmakokinetyce substancji leczniczych, można wyrazić matematycznie. Jest to konieczne przy planowaniu schematów do użytku klinicznego. leki... Na podstawie danych farmakokinetycznych opracowywane są zasady racjonalnego doboru i dawkowania tych ostatnich. Jednocześnie wraz z tymi obliczeniami wymagane jest stałe monitorowanie kliniczne działania leku, ponieważ badania farmakokinetyczne jedynie uzupełniają tę kontrolę i pozwalają na wyciągnięcie bardziej obiektywnych wniosków.
Eliminacja większości substancji leczniczych odbywa się zgodnie z kinetyką wykładniczą, a mianowicie w taki sposób, że za każdy równy okres czasu z organizmu znika stała część całkowitej ilości podanej substancji leczniczej. W większości przypadków szybkość, z jaką lek znika z organizmu, znajduje odzwierciedlenie w odpowiedniej szybkości spadku poziomu leku w osoczu.
Stężenie leków w płynach biologicznych określa się za pomocą chromatografii cieczowej lub gazowo-cieczowej, testu radioimmunologicznego lub analizy enzymatycznej, polarografii lub spektrofotometrii. Wielokrotne oznaczanie stężeń leku we krwi w trakcie leczenia nazywa się MONITOROWANIEM TERAPEUTYCZNYM. W tym celu czasami używa się śliny, która jest bezbiałkowym ultrafiltratem krwi.
Na podstawie uzyskanych wartości sporządzany jest wykres, na odciętej którego zaznaczony jest czas pobierania próbki, a na rzędnej stężenie leku w próbce biologicznej (najczęściej w osoczu krwi) w odpowiednich jednostkach. Otrzymana krzywa charakteryzuje procesy farmakokinetyczne zachodzące z lekiem. Tak więc po pojedynczym wstrzyknięciu dożylnym stężenie leku w osoczu spada wykładniczo. Szybkość procesu wykładniczego można scharakteryzować jako stałą szybkości (K), odzwierciedlającą zmianę stężenia w jednostce czasu lub w okresie procesu półwykładniczego (oznaczoną jako T 1/2 lub t / 2). Okres ten jest równy czasowi zakończenia procesu o 50%.
Eliminację leków z organizmu można ocenić na podstawie okresu półtrwania lub okresu półtrwania, okresu półtrwania, okresu półtrwania, który jest definiowany jako czas do obniżenia się stężenia leku we krwi o 50% podanej ilości leku lub eliminacja 50% biodostępnej ilości leku.
Termin „OKRES PÓŁELIMINACYJNY” jest bardziej skuteczny niż „OKRES PÓŁELIMINACYJNY”, ponieważ leki są nie tylko wydalane, ale także ulegają biotransformacji. Okres półtrwania eliminacji można określić na podstawie wykresu stężenie-czas, mierząc przedział czasu, w którym dowolne stężenie substancji na krzywej zmniejszyło się o połowę.
Praktycznie ważne jest, aby pamiętać, że w jednym okresie półtrwania
50% leku jest wydalane z organizmu, 75% przez dwa okresy, 90% przez trzy okresy, 94% przez cztery.
Ponieważ całkowite wyeliminowanie typu wykładniczego wymaga czasu dłuższego niż cztery (4) okresy półtrwania, to przy wielokrotnym podawaniu leku w krótszych odstępach czasu odnotowuje się jego kumulację (akumulację). Szacuje się, że do osiągnięcia plateau stężenia, to znaczy stałego stężenia leku w osoczu, potrzeba około czterech okresów biologicznego okresu półtrwania leku.
Ważne jest, aby zmniejszenie eliminacji leku prowadziło do wydłużenia biologicznego okresu półtrwania i przedłużenia działania leku.
W przypadku niektórych leków efekt farmakologiczny może być dłuższy niż sugerowałby t/2. W związku z tym leki takie jak hormon wzrostu, anaprilin można podawać w odstępach dłuższych niż ich T / 2.
Aby uniknąć niebezpiecznego wzrostu poziomu leku w osoczu u pacjentów ze zmniejszoną eliminacją w przypadku niewydolności wątroby, nerek lub układu sercowo-naczyniowego, jego dawki podtrzymujące należy zmniejszać albo zmniejszając każdą dawkę, albo wydłużając odstępy między podaniami proporcjonalnie do wydłużenie ich biologicznego okresu półtrwania....
BIOLOGICZNA DOSTĘPNOŚĆ LEKÓW
Aby zapewnić efekt terapeutyczny, substancja lecznicza musi zostać dostarczona do tych narządów lub tkanek, w których odbywa się jej specyficzne działanie (w biofazie). Przy wstrzyknięciu donaczyniowym lek natychmiast i całkowicie dostaje się do krwioobiegu. W przypadku innych dróg podania (doustnie, domięśniowo, podskórnie itp.) przed dostaniem się do krwiobiegu lek musi przejść przez szereg biologicznych błon komórkowych (błona śluzowa żołądka, komórki wątroby, mięśnie itp.) i dopiero potem część wejdzie do krążenia ogólnoustrojowego. Działanie leku w dużej mierze zależy od tego, ile podanej dawki leku dostanie się do krążenia ogólnoustrojowego. Wskaźnik ten charakteryzuje biodostępność środka (F). Zatem w istocie biodostępność leku odzwierciedla jego stężenie w receptorach, to znaczy we krwi i tkankach ciała po wchłonięciu. Naturalnie biodostępność tego samego środka będzie różna dla każdego pacjenta. Oczywiście przy dożylnym podaniu leku jego biodostępność wynosi około 100%, a przy innych drogach podawania biodostępność prawie nigdy nie osiąga 100%.
Rozróżnij BIODOSTĘPNOŚĆ ABSOLUTNĄ I WZGLĘDNĄ. Biodostępność bezwzględna to stosunek wchłoniętego leku podczas podawania pozanaczyniowego do jego ilości po podaniu dożylnym.
Ważnym wskaźnikiem jest WZGLĘDNA BIODOSTĘPNOŚĆ, która określa względny stopień wchłaniania leku z leku badanego i z leków referencyjnych. Innymi słowy, względna biodostępność jest określana dla różnych serii leków, w przypadku zmiany leków
technologia produkcji, dla leków produkowanych przez różnych producentów w różnych postaciach dawkowania. Do określenia względnej biodostępności można wykorzystać dane dotyczące poziomu substancji leczniczej we krwi lub jej wydalania z moczem po jednorazowym lub wielokrotnym podaniu. Termin ten jest ważny przy porównywaniu 2 leków ze sobą.
Biodostępność porównawcza tych samych leków wytwarzanych przez różne firmy (przykład: kokarboksynaza pochodzenia polskiego i wyprodukowana w Dniepropietrowsku) określana jest poprzez porównanie równoważników chemicznych, biologicznych i terapeutycznych.
RÓWNOWAŻNOŚĆ CHEMICZNA to przypadek nie tylko w lekach wzór chemiczny leki, ale także koincydencja izomerii, przestrzennej konfiguracji atomów w cząsteczce leku.
RÓWNOWAŻNOŚĆ BIOLOGICZNA oznacza to samo, równe stężenie substancja aktywna we krwi podczas przyjmowania leku z różnych firm.
Wreszcie RÓWNOWAŻNOŚĆ TERAPEUTYCZNA oznacza ten sam, równoważny efekt terapeutyczny.
Jeśli wymienione 3 cechy są takie same, mówi się, że leki mają równą biodostępność (biodostępność). Obecnie istnieje wiele przykładów, że podobne leki nie są biologicznie równoważne ze względu na różnice w biodostępności. Praktyk powinien o tym pamiętać, zwłaszcza przy przenoszeniu pacjenta z jednego leku na podobny lek inna firma.
Oczywiście na wszystkie te pytania może odpowiedzieć tylko nowa nauka - a mianowicie FARMAKOLOGIA KLINICZNA. Jest to niezależna nauka z własnymi celami przedmiotowymi i badawczymi. Dlaczego wyróżniał się jako niezależny podmiot? Przede wszystkim dlatego, że, jak się okazało, nie wszystko da się zbadać w eksperymencie na zwierzętach. Na przykład, procesy mentalne, które są wysoce charakterystyczne tylko dla człowieka.
Szybki rozwój przemysłu farmaceutycznego doprowadził do powstania ogromnej liczby leków. Pojawiła się lawina leków, tworząc rodzaj leczniczej dżungli. Obecna sytuacja bardzo utrudnia wybór właściwe narzędzie nawet w jednej grupie leków uniemożliwia lekarzowi zorientowanie się na lek, który jest optymalny dla konkretnego pacjenta. Farmakologia kliniczna pomaga odpowiedzieć na wszystkie te pytania.
Przykładem jest możliwość wyboru leku na choroby kolagenowe (choroby tkanki łącznej, reumatoidalne zapalenie stawów, reumatyzm, toczeń rumieniowaty układowy itp.). Z jednym
boki - kwas acetylosalicylowy (aspiryna), ale jednocześnie istnieją inne nowoczesne nie-narkotyczne środki przeciwbólowe, które w porównaniu z aspiryną mają szereg zalet: naproksen, piroksykam itp.
Co jest lepsze, który lek będzie bardziej odpowiedni dla danego pacjenta, który daje najsilniejszy efekt terapeutyczny? Farmakologia kliniczna pomaga odpowiedzieć na te pytania.
Główne zadania farmakologa klinicznego to:
1) Wybór leków do leczenia konkretnego bólu
2) Ustalenie najbardziej odpowiednich dla niego produktów leczniczych
formy i tryb ich stosowania.
3) Wybór drogi podania leku.
4) Monitorowanie działania leku.
W tym celu zainstalowane są czujniki, które dają na monitorze stały obraz stężenia leku we krwi. Badane są wszystkie inne aspekty farmakokinetyki.
5) Badanie działań niepożądanych i skutków ubocznych leków, ich eliminacji, a także badanie konsekwencji interakcji leków u danego pacjenta.
6) Transfer zgromadzonej wiedzy poprzez szkolenia.
7) Organizacja laboratorium i usługi informacyjne oraz porady dotyczące planowania badań (WHO, 1971).
FARMAKODYNAMIKA (PD) to gałąź farmakologii, która bada
1) mechanizmy działania (czyli istota procesów interakcji z receptorami tkankowymi, komórkowymi lub subkomórkowymi - specyficznymi lub niespecyficznymi) 1.
1 02) efekty farmakologiczne (to znaczy zawartość i zmiany w działaniu leku w zależności od wieku, płci pacjenta, charakteru i przebiegu choroby, współistniejącej patologii), a także 3) lokalizacji leku akcja. Krótko mówiąc, PD można zdefiniować jako dział farmakologii, który bada wpływ leków na organizm.
Zwykle mechanizm działania leku bada się w eksperymentach na zwierzętach, ponieważ są one prawie zawsze takie same u zwierząt i ludzi. Znajomość mechanizmu działania leku pozwala lekarzowi inteligentnie dobrać lek niezbędny do leczenia.
Istnieje wiele mechanizmów działania leków, ale wszystkie można warunkowo zredukować do 2 grup.
Pierwsza grupa mechanizmów związana jest z przypadkami, w których leki działają na określone receptory – czyli są to MECHANIZMY RECEPTORÓW.
Druga grupa mechanizmów związana jest z lekami, które ze względu na ich: fizyczne i chemiczne właściwości nie działają przez receptory. Tu przede wszystkim można wskazać wpływ leków na określone enzymy, ich fizykochemiczny wpływ na błony komórkowe oraz bezpośrednie oddziaływanie chemiczne z substancjami komórkowymi.
Przykładem mechanizmów niereceptorowych jest
przypadek z lekami do znieczulenia, powiedzmy z fluorotanem. Jest doskonałym rozpuszczalnikiem dla tłuszczów, dlatego działa przede wszystkim na błony komórek nerwowych, wywołując efekt farmakologiczny – znieczulenie.
Przyjrzyjmy się głównym, najczęściej spotykanym receptorom i mechanizmom działania leków.
W ujęciu farmakologicznym receptory to funkcjonalne biochemiczne struktury błon wielkocząsteczkowych, które są selektywnie wrażliwe na działanie niektórych związków chemicznych, aw naszym przypadku na działanie leków. Badania ostatnie lata wykazali, że receptorami farmakologicznymi są białka lub enzymy (białka G to pojedynczy łańcuch peptydowy 7 domen) - w tym podstawowa różnica z receptorów morfologicznych.
Selektywna wrażliwość leku na receptor oznacza, że lek może w pierwszej kolejności wiązać się z receptorem, czyli ma do niego powinowactwo lub powinowactwo. Innymi słowy, powinowactwo lub powinowactwo odnosi się do zdolności leku do wiązania się z receptorem.
Powinowactwo lub powinowactwo odzwierciedla stałe kinetyczne wiążące lek, receptor i odpowiedź na response Poziom molekularny... Interakcja substancji leczniczych z receptorem prowadzi do pojawienia się szeregu biochemicznych i zmiany fizjologiczne w ciele, które wyrażają się w takim czy innym efekcie.
Drugą cechą substancji leczniczej jest jej zdolność do wywoływania odpowiedzi farmakologicznej, efektu po interakcji z receptorem. Ta zdolność jest określana jako samoistna aktywność leku lub jego skuteczność. Do pewnego stopnia odpowiedź biologiczna jest regulowana poprzez zmianę liczby receptorów i ich wrażliwości.
W toku ewolucji powstały receptory wrażliwe na różne endogenne regulatory. Zgodnie z teorią receptorową mechanizm działania leków polega na zmianie tempa funkcjonowania określonych układów organizmu, gdy na receptory działają naturalne mediatory lub substancje egzogenne.
Leki, których działanie związane jest z bezpośrednim pobudzeniem lub wzrostem funkcjonalność(zdolności) receptorów nazywane są AGONIKAMI, a substancje zakłócające działanie określonych agonistów nazywane są ANTAGONISTAMI. Innymi słowy, jeśli lek ma obie cechy (to znaczy zarówno powinowactwo, jak i aktywność wewnętrzną), to jest agonistą. Dlatego agonista jest substancją o wysokim powinowactwie do receptora i wysokiej aktywności wewnętrznej. Jeśli substancja ma zdolność wiązania się tylko z receptorem (czyli ma powinowactwo), ale jednocześnie nie jest w stanie wywołać efektów farmakologicznych, to powoduje blokadę receptora i nazywana jest antagonistą.
Leki, które mają takie samo powinowactwo do receptora jak agonista lub słabsze, ale mają mniej wyraźny
działania nazywane są częściowymi agonistami lub agonistami-antagonistami. Leki te, stosowane jednocześnie z agonistami, zmniejszają działanie tych ostatnich ze względu na ich zdolność do zajmowania receptora.
Przykład: atropina – ma wyższą aktywność niż acetylocholina (mediator endogenny). Atropina oddziałuje z receptorami, ale ponieważ nie ma aktywności wewnętrznej, nie wywoła efektu fizjologicznego. Ze względu na większe powinowactwo do receptora w porównaniu do acetylocholiny będzie on zakłócał działanie agonisty, czyli acetylocholiny, a zatem będzie jego antagonistą.
Substancje lecznicze mogą działać podobnie lub przeciwnie do mediatorów endogennych. Jeśli substancja lecznicza działa jak mediator (acetylocholina, norepinefryna itp.), taka substancja nazywa się MIMETIC. Mim - rdzeń „mim”, pantomima, mimikra. Stąd cholinomimetyk, agonista adrenergiczny.
Lek, który zapobiega interakcji mediatora z receptorem, nazywany jest blokerem (antycholinergiczny, bloker adrenergiczny, bloker histaminy itp.).
W literaturze można znaleźć termin „lityczny” (liza – rozpuszczanie, proces fizyczny). Termin jest dość stary, ale czasami jest używany (antycholinergiczny, adrenolityczny). Zatem terminy „lityczny” i „bloker” są używane jako synonimy.
W praktyce lekarskiej coraz więcej szerokie zastosowanie znajduje jednoczesne powołanie kilku leków. Jednocześnie mogą wchodzić ze sobą w interakcje, zmieniając nasilenie i charakter głównego efektu, czas jego trwania lub osłabiając skutki uboczne i toksyczne. W związku z tym specjalna sekcja farmakodynamiki poświęcona jest INTERAKCJI LEKÓW, która jest klasyfikowana w następujący sposób. Rozróżnij interakcję FARMAKOLOGICZNĄ i interakcję FARMACEUTYCZNĄ.
Interakcje farmaceutyczne wiążą się z niezgodnością farmaceutyczną leków podczas ich wytwarzania lub przechowywania, a także podczas mieszania w jednej strzykawce. Jednocześnie zmniejsza się lub zanika dotychczasowa aktywność farmakologiczna leków, a czasami pojawiają się nawet nowe, toksyczne właściwości.
Oddziaływanie farmakologiczne leków wiąże się ze zmianami ich farmakokinetyki, farmakodynamiki lub opiera się na oddziaływaniach chemicznych i fizykochemicznych w środowiskach organizmu. W tym przypadku leki mogą wchodzić ze sobą w interakcje na każdym etapie ich przechodzenia przez organizm pacjenta: podczas wchłaniania, w fazie transportu, w procesie metabolizmu, a także wydalania (interakcja farmakokinetyczna).
Interakcja farmakodynamiczna odzwierciedla zmianę wywołaną przez każdy lek z osobna procesy związane z realizacją efektu. Innymi słowy, farmakodynamiczny rodzaj interakcji opiera się na osobliwościach zmian w mechanizmach i lokalizacji działania stosowanych leków, ich głównych efektach. Jeżeli interakcja zachodzi na poziomie receptora, to dotyczy głównie agonistów i antagonistów różnych typów receptorów. W takim przypadku jedna substancja lecznicza może wzmocnić lub osłabić działanie innej. Jeśli ve . lecznicza
Substancje działają w stosunku do efektu jednokierunkowo – są to leki synergiczne (syn – razem, ergo – praca). Tym samym synergizmowi towarzyszy wzrost efektu końcowego. Zazwyczaj leki te działają na te same receptory. Istnieją 2 warianty synergii:
1) Efekty są takie same na zasadzie prostej sumy. Podsumowując (lub dodatek, - łac. - additio - dodatek). Efekt obserwuje się po prostu dodając efekty każdego ze składników. Na przykład w ten sposób oddziałują leki znieczulające (podtlenek azotu + fluorotan). Wariant efektu addytywnego jest podobny przy jednoczesnym stosowaniu aspiryny i analginy. Dlaczego musisz to wiedzieć? Jeśli pacjent jest zmuszony do zażywania aspiryny długi czas, należy wziąć pod uwagę, że Aspiryna działa wrzodziejąco, to znaczy powoduje owrzodzenie błony śluzowej przewodu pokarmowego, a Analgin ma taki niepożądany efekt, jak hamowanie hematopoezy. Biorąc pod uwagę addytywne działanie przeciwbólowe, możliwe jest zmniejszenie bez znaczącego ryzyka jego wystąpienia, znaczne zmniejszenie dawki obu leków przyjmowanych przez pacjenta.
2) Druga wersja synergii to wzmocnienie lub wzmocnienie efektu. Ta opcja ma miejsce, gdy przy wprowadzeniu dwóch substancji łączny efekt przekracza sumę efektów obu środków. Przykładem jest interakcja leków przeciwpsychotycznych (aminazyna) i środków znieczulających, interakcja antybiotyków i przeciwdrobnoustrojowych sulfonyloamidów.
Czasami wyróżnia się trzeci (3) wariant synergizmu - uczulenie. Uczulenie - gdy jeden lek w minimalnej dawce wzmacnia działanie innego w ich połączeniu (zastosowanie małych dawek insuliny w połączeniu z KCl zwiększa poziom przenikania potasu do komórek).
Oprócz synergii występuje zjawisko antagonizmu. Zdolność jednej substancji do zmniejszania działania innej w takim czy innym stopniu nazywa się ANTAGONIZM, co oznacza, że w tym przypadku jeden lek zapobiega działaniu drugiego.
Istnieją antagonizmy fizyczne, chemiczne i fizjologiczne. Ten widok interakcje są najczęściej stosowane w przypadku przedawkowania lub ostrego zatrucia lekami. Przykładem FIZYCZNEGO antagonizmu może być zdolność adsorbentów do utrudniania wchłaniania substancji z przewód pokarmowy(węgiel aktywny, który adsorbuje truciznę na swojej powierzchni; cholestyramina).
Ilustracją oddziaływania CHEMICZNEGO może być tworzenie się kompleksonów (jony niektórych metali ciężkich - rtęci, ołowiu - wiążą penicyloaminę, EDTA) lub tak to oddziałuje kwas chlorowodorowyżołądek i wodorowęglan sodu (zasady).
Antagonizm FIZJOLOGICZNY wiąże się z interakcjami leków na poziomie receptorów, których charakter został już omówiony powyżej.
Analogicznie do synergizmu rozróżnia się antagonizm BEZPOŚREDNI (gdy oba związki leków działają na te same receptory) i POŚREDNI (inna lokalizacja działania leku). Z kolei bezpośredni antagonizm jest KONKURENCYJNY i NIE
KONKURENCYJNY. Dzięki antagonizmowi konkurencyjnemu lek wchodzi w relację konkurencyjną z naturalnymi regulatorami (mediatorami) o miejsca wiązania w określonych receptorach. Blokadę receptora wywołaną przez konkurencyjnego antagonistę można złagodzić dużymi dawkami agonisty lub naturalnego mediatora.
Antagonizm niekonkurencyjny to sytuacja, w której lek nie może wyprzeć naturalnego mediatora z receptora, ale tworzy z nim wiązania kowalencyjne (mediator).
PUNKTY INTERAKCJI LEKÓW Główna
masa receptorów znajduje się po zewnętrznej i wewnętrznej stronie błony komórkowej i jej organelli. Do najczęstszych punktów interakcji leków należą: 1) mediatory i receptory hormonalne; 2) Faza ATP pompy Na / K, Ca, K i Na - kanały wewnątrzbłonowe.
To ostatnie udowadnia po raz kolejny, że leki działają na dostępne kluczowe mechanizmy reakcji biologicznych, czyli na procesy zdeterminowane filogenetycznie, a nie poprzez tworzenie nowych reakcji.
Interakcja leków z receptorem zachodzi na poziomie procesów chemicznych lub fizykochemicznych. Najczęściej charakter reakcji, jej siła, odwracalność i czas trwania wynikają z właściwości połączenia leku z receptorem. Siła wiązania zależy od odległości oddziaływania elektrostatycznego między dwoma atomami. Z reguły charakter interakcji jest złożony, może obejmować: Różne rodzaje komunikacja, która jest określona przez komplementarność leku i receptora, stopień ich zbieżności ze sobą.
Najsłabsze wiązania to van der Waals (określają specyfikę oddziaływania substancji z układami reaktywnymi). W większości przypadków między lekiem a receptorem powstają wiązania jonowe (odwracalne).
RODZAJE DZIAŁANIA LEKÓW
1) EFEKT LOKALNY – działanie substancji występujące w miejscu jej aplikacji. Przykład: zastosowanie środków miejscowo znieczulających – wprowadzenie roztworu dikainy do jamy spojówki. Zastosowanie 1% roztworu nowokainy podczas ekstrakcji
ząb. Ten termin (działanie lokalne) jest nieco arbitralny, ponieważ prawdziwie lokalne działanie obserwuje się niezwykle rzadko, ponieważ substancje mogą być częściowo wchłaniane lub mieć efekt odruchowy.
2) DZIAŁANIE ODBLASKOWE - wtedy lek działa na drogi odruchu, to znaczy wpływa na zewnętrzne lub interoreceptory, a efekt objawia się zmianą stanu odpowiednich ośrodków nerwowych lub narządów wykonawczych. Tak więc stosowanie plastrów musztardowych do patologii układu oddechowego poprawia odruchowo ich trofizm (niezbędny olejek musztardowy stymuluje zewnętrzne receptory skóry). Lek cytyton (analeptyk oddechowy) działa pobudzająco na chemoreceptory kłębuszka szyjnego i poprzez odruchową stymulację ośrodka oddychania zwiększa objętość i częstotliwość oddychania. Innym przykładem jest zastosowanie amoniaku do omdlenia (amoniak), który odruchowo poprawia
krążenie mózgowe i toniczne ośrodki witalne.
3) DZIAŁANIE RESORPCYJNE - wtedy działanie substancji rozwija się po jej wchłonięciu (resorpcja - wchłanianie; łac. - resorbeo - wchłaniam), dostaniu się do ogólnego krwiobiegu, a następnie do tkanek. Działanie resorpcyjne zależy od drogi podania leku i jego zdolności do przenikania barier biologicznych. Jeśli substancja oddziałuje tylko z funkcjonalnie jednoźródłowymi receptorami o określonej lokalizacji i nie wpływa na inne receptory, działanie takiej substancji nazywa się SELEKTYWNYM. Tak więc niektóre substancje kurarypodobne (środki zwiotczające mięśnie) dość selektywnie blokują receptory cholinergiczne płytki końcowej, powodując rozluźnienie mięśni szkieletowych. Działanie leku prazosin wiąże się z selektywnym blokowaniem postsynaptycznych receptorów alfa-adrenergicznych, co ostatecznie prowadzi do zmniejszenia ciśnienie krwi... Podstawą selektywności działania leków (selektywność) jest powinowactwo (powinowactwo) substancji do receptora, które określa obecność pewnych grup funkcyjnych w cząsteczce tych substancji i ogólna strukturalna organizacja substancji , który jest najbardziej odpowiedni dla interakcji z tymi receptorami, czyli KOMPLETNOŚĆ.
OGÓLNA CHARAKTERYSTYKA WPŁYWU LEKÓW NA ORGANIZM
Pomimo obfitości leków, wszystkie ogromne
wywołane przez nie w organizmie, mają pewną cechę wspólną i
jednolitość. W oparciu o koncepcję szybkości reakcji istnieje 5
rodzaje zmian
spowodowane przez środki farmakologiczne (N.V. Vershinin):
1) tonowanie (zwiększenie funkcji do normy);
2) podniecenie (zwiększona funkcja powyżej normy);
3) działanie uspokajające (uspokajające), czyli obniżające ulepszona funkcja do normy;
4) ucisk (spadek funkcji poniżej normy);
5) paraliż (zakończenie funkcji). Ilość toniku i koszyka
efekt przebudzenia nazywany jest akcją przebudzenia.
GŁÓWNE EFEKTY LEKÓW
Przede wszystkim są:
1) efekty fizjologiczne, gdy leki powodują zmiany, takie jak wzrost lub spadek ciśnienia krwi, częstości akcji serca itp.;
2) biochemiczne (podwyższony poziom enzymów we krwi, glukozy
Shl itp.). Ponadto istnieją BASIC (lub główne) i
WTÓRNE (drobne) skutki leków. PODSTAWOWY EF
FAKT to ten, na którym lekarz opiera swoje kalkulacje w leczeniu tego (!) Pacjenta (leki przeciwbólowe – o działaniu znieczulającym, przeciwnadciśnieniowe – na obniżenie ciśnienia itp.).
ZNACZĄCE lub inne niż główne skutki, dodatkowe w przeciwnym razie te, które są nieodłączne to narzędzie, ale których opracowanie u danego pacjenta nie jest konieczne (nienarkotyczne leki przeciwbólowe – oprócz działania przeciwbólowego wywołują działanie przeciwgorączkowe itp.). Efekty inne niż główne mogą obejmować efekty POŻĄDANE i NIEPOŻĄDANE (lub UBOCZNE).
Przykład. Atropina - rozluźnia mięśnie gładkie wewnętrzne
organy. Jednocześnie jednak jednocześnie poprawia przewodzenie w węźle AV serca (z blokiem serca), zwiększa średnicę źrenicy itp. Wszystkie te efekty należy w każdym przypadku rozpatrywać indywidualnie.
CZYNNIKI WPŁYWAJĄCE NA WARTOŚĆ DZIAŁANIA NARKOTYKÓW
1) Przede wszystkim należy pamiętać o czynnikach farmakokinetycznych właściwych dla każdego leku. Zostało to już wspomniane powyżej, przypomnę tylko, że mówimy o szybkości jego wchłaniania lub wchłaniania, biotransformacji, wydalania (lek, lek).
2) Druga grupa czynników to czynniki fizjologiczne.
a) Wiek. Rzeczywiście, wszyscy doskonale zdają sobie sprawę, że wraz z wiekiem zmienia się wrażliwość pacjenta na leki. Wyróżniali się nawet pod tym względem:
Farmakologia okołoporodowa;
Farmakologia dziecięca;
Farmakologia geriatryczna;
Farmakologia rozrodu;
b) Waga pacjenta. Wiadomo, że im większa masa, tym wyższa
dawka. Dlatego leki są dawkowane w (mg / kg).
c) Płeć. Ujawnił różną wrażliwość u mężczyzn i kobiet
niektóre substancje, np. nikotyna, alkohol itp.,
co tłumaczy się różnicą w metabolizmie, różnicą w swoistości
masa tkanki tłuszczowej itp.
c) Stan organizmu. Wpływ leków na organizm po su
ćwiczenia fizyczne będą inne niż bez niego.
mi) Rytmy biologiczne(dzienna, miesięczna, sezonowa, roczna
vye, a teraz nawet populacja) mają najpoważniejsze
wpływ na działanie leków w organizmie. 3) Fakty patologiczne
ry (na przykład poziom aktywności hormonalnej). Więc z ba
toksyczne dawki morfiny są łatwiej tolerowane, ale wzrasta wrażliwość mięśnia sercowego na adrenalinę. 1 0 Wpływ glikozydów nasercowych na krążenie krwi przejawia się tylko na tle niewydolności serca. Wpływ leków zmienia się znacząco wraz z hipo- i hipertermią, chorobami zakaźnymi, zmianą stanu funkcjonalnego ośrodkowego układu nerwowego itp.).
4) Czynniki genetyczne. Wiadomo, że brak enzymu dehydrogenazy glukozo-6-fosforanowej (G-6-FDG) w talasenii uniemożliwia przepisywanie leków przeciwmalarycznych, takich jak prymachina. Niedobór enzymu butyrylocholinesterazy we krwi, który występuje u co 2500 osób, jest przyczyną przedłużonego zwiotczenia mięśni po podaniu ditiliny.
5) Sugestia pacjentów lub efekt placebo. Pod tym względem działanie przeciwdławicowe leków placebo, na przykład, osiąga 40%, a do 81% efektu placebo wynika z drogi wstrzyknięcia podawania leku. Prawdopodobnie dlatego używam preparaty witaminowe, toniki, środki uspokajające są w dużej mierze spowodowane tym efektem.
6) Dawka leku. O działaniu leków w bardzo dużym stopniu decyduje ich dawka. Dawka odnosi się do ilości substancji leczniczej
pojedyncza dawka (zwykle określana jako pojedyncza dawka). Dawka leku zależy nie tylko od skuteczności leczenia, ale także od bezpieczeństwa pacjenta. Pod koniec XVIII wieku William Withering napisał: „Trucizna w małych dawkach jest najlepszym lekarstwem; użytecznym lekarstwem w zbyt dużej dawce jest trucizna”. Jest to tym bardziej prawdziwe w naszych czasach, kiedy do praktyki medycznej wprowadzono niezwykle aktywne leki, których dawki mierzone są w ułamkach miligrama.
Podaj dawkę w gramach lub ułamkach grama. W celu dokładniejszego dawkowania leków ich ilość oblicza się na 1 kg masy ciała (lub na 1 metr kwadratowy powierzchni ciała), na przykład 1 mg / kg; 1 μg / kg itp. Lekarz musi być zorientowany nie tylko na dawkę obliczoną dla pojedynczej dawki (pro dosi), ale także na dawkę dzienną (pro die).
Minimalne dawki, w których leki wywołują początkowy efekt biologiczny (terapeutyczny), nazywane są progowymi lub minimalnymi skutecznymi (leczniczymi) dawkami. W medycynie praktycznej najczęściej stosuje się średnie dawki terapeutyczne, w których leki mają niezbędny optymalny efekt farmakoterapeutyczny. Jeśli podczas przepisywania ich pacjentowi efekt nie jest wystarczająco wyraźny, dawkę zwiększa się do najwyższej dawki terapeutycznej. Wyższe dawki terapeutyczne mogą być pojedyncze i dzienne. Najwyższa pojedyncza dawka to maksymalna ilość leku, którą można podać jednorazowo bez szkody dla pacjenta. Dawki te są stosowane rzadko, w skrajnych przypadkach (w nagłych, nagłych sytuacjach). Średnie dawki terapeutyczne wynoszą zwykle 1/3-1/2 najwyższej pojedynczej dawki.
Wyższe dawki terapeutyczne trujących i silne substancje są podane w Farmakopei Państwowej ZSRR. W niektórych przypadkach, na przykład przy stosowaniu środków chemioterapeutycznych, dawka leku jest wskazana dla przebiegu leczenia (dawka kursu). Jeśli zajdzie potrzeba szybkiego wytworzenia wysokiego stężenia leku w organizmie (posocznica, niewydolność sercowo-naczyniowa), należy zastosować pierwszą dawkę, tzw. dawkę nasycającą, która przewyższa wszystkie kolejne. Istnieją również dawki toksyczne (niebezpieczne skutki) i śmiertelne.
Ważne jest, aby lekarz znał jeszcze jedną cechę - a mianowicie pojęcie zakresu terapeutycznego działania leku. Zakres działania terapeutycznego rozumiany jest jako odległość, zakres od minimalnej dawki terapeutycznej do minimalnej dawki toksycznej. Oczywiście im większa odległość, tym lek jest bezpieczniejszy.
1/20 dawki x liczba lat dziecka.
Aby ilościowo scharakteryzować i ocenić skuteczność nowego środka farmakologicznego, zwykle stosuje się dwa standardowe porównania - albo z placebo, albo z lekiem analogowym.
giczny typ działania, który jest jednym z najskuteczniejszych środków w tej grupie.
Placebo (atrapa) to obojętna substancja w postaci dawkowania, która naśladuje określony środek farmakologiczny lub lek. Stosowanie placebo jest konieczne w przypadku: a) efektu domniemania, wpływu osobowości, oczekiwań i stronniczość przez pacjenta lub badacza; b) samoistne zmiany w przebiegu choroby, objawy, a także zjawiska niezwiązane z leczeniem.
Placebo to łaciński termin oznaczający „mogę cię zadowolić”.
Efekt placebo to efekt wywołany nie specyficznymi właściwościami farmakodynamicznymi leku dla danej patologii, ale FAKTEM UŻYWANIA leków, który wpływa na psychikę. Leki placebo są zwykle farmakologicznie obojętne i zawierają substancje nieaktywne, takie jak skrobia czy laktoza. Placebo stosuje się w badaniach klinicznych w celu określenia wpływu sugestii ze strony zarówno pacjenta, jak i lekarza, zwłaszcza jeśli badane mają być leki przeznaczone do leczenia. astma oskrzelowa, nadciśnienie, dusznica bolesna, choroba niedokrwienna serca. W takich przypadkach lek placebo nie powinien być zabarwiony ani w inny sposób właściwości fizyczne(zapach, smak, kształt) różnią się od aktywnego leku. Placebo jest bardziej skuteczne, gdy zarówno lekarz, jak i pacjent są o nim słabo poinformowani.
PRZYKŁAD. Na choroba niedokrwienna choroba serca (IHD), jeśli jednej grupie pacjentów z IHD przepisano aktywny lek, a drugiej - placebo, wówczas u 40% pacjentów z drugiej grupy napady dusznicy bolesnej zostają zatrzymane.
Bardzo wyraźny efekt placebo (do 81%) obserwuje się przy drodze wstrzyknięcia jego podania. Mikstury i pigułki są mniej skuteczne.
Termin FARMAKOTERAPIA (FT) jest często używany w literaturze dotyczącej narażenia pacjenta na lek. Farmakoterapia to dział farmakologii, który bada farmakoterapię pacjenta.
Wyróżnić następujące typy farmakoterapia:
1) ETIOTROPOWE - idealny widok farmakoterapia. Ten rodzaj FT ma na celu wyeliminowanie przyczyny choroby. Przykładami etiotropowego PT może być leczenie pacjentów zakaźnych środkami przeciwdrobnoustrojowymi (penicylina benzylowa na paciorkowcowe zapalenie płuc), stosowanie odtrutek w leczeniu pacjentów z toksycznymi
Substancje. 2) FARMAKOTERAPIA PATOGENETYCZNA - skierowana
wyeliminować lub
tłumienie mechanizmów rozwoju choroby. Większość obecnie stosowanych leków należy konkretnie do grupy leków patogenetycznych FT. Leki przeciwnadciśnieniowe, glikozydy nasercowe, przeciwarytmiczne, przeciwzapalne, psychotropowe i wiele innych mają działanie terapeutyczne poprzez hamowanie odpowiednich mechanizmów rozwoju choroby.
3) TERAPIA OBJAWOWA - mająca na celu wyeliminowanie lub
ograniczenie indywidualnych objawów choroby. Leki objawowe obejmują środki przeciwbólowe, które nie wpływają na przyczynę ani mechanizm choroby. Leki przeciwkaszlowe są również dobrym przykładem środków objawowych. Czasami te środki zaradcze (eliminacja zespół bólowy z zawałem mięśnia sercowego) może mieć znaczący wpływ na przebieg głównego proces patologiczny a jednocześnie odgrywają rolę środków terapii patogenetycznej.
4) FARMAKOTERAPIA ZAMIENNA stosowana jest w przypadku niedoboru naturalnych składników odżywczych. Środki terapii zastępczej obejmują preparaty enzymatyczne (pankreatyna, panzinorm itp.), leki hormonalne (insulina na cukrzycę, tarczyca na obrzęk śluzowaty), preparaty witaminowe (witamina D, na przykład na krzywicę). Leki do terapii substytucyjnej, bez eliminowania przyczyn choroby, mogą zapewnić normalne istnienie organizmu przez wiele lat. To nie przypadek, że tak poważna patologia jak cukrzyca- jest uważany za szczególny styl życia wśród Amerykanów.
5) TERAPIA PREWENCYJNA prowadzona jest w celu zapobiegania chorobom. Niektóre są zapobiegawcze środki przeciwwirusowe(na przykład podczas epidemii grypy - remantadyna), środki dezynfekujące i wiele innych. Profilaktyczne FT można również uznać za stosowanie leków przeciwgruźliczych, takich jak izonizyd. Dobry przykład prowadzenie terapii zapobiegawczej to stosowanie szczepionek.
CHEMIOTERAPIA należy odróżnić od farmakoterapii. Jeśli FT zajmuje się dwoma uczestnikami procesu patologicznego, a mianowicie lekiem i makroorganizmem, to podczas chemioterapii jest już 3 uczestników: lek, makroorganizm (pacjent) i czynnik sprawczy choroby.
Mówiąc o dawkach, wskazaliśmy przede wszystkim na dawki alopatyczne, a nie homeopatyczne. Dlatego kilka słów o HOMEOPATII. Termin „homeopatia” wywodzi się z dwóch greckich słów: homois – podobny i pathos – cierpienie, choroba. Dosłownie homeopatia jest tłumaczona jako podobna, podobna choroba. Założyciel homeopatii, niemiecki naukowiec Samuel Hahnemann, w swojej słynnej książce „Organon sztuki medycznej lub podstawowa teoria leczenia homeopatycznego” z powrotem w początek XIX wieku (1810) nakreślił podstawowe zasady tej nauki. Istnieje kilka ich zasad, ale 2 z nich są podstawowe:
1) Jest to prawo podobieństwa, które mówi, że leczenie chorób musi odbywać się podobnymi, podobnymi środkami. Zgodnie z tą zasadą Hahnemann radzi „naśladować naturę, która czasami leczy przewlekłą chorobę poprzez inną współistniejącą chorobę”. Dlatego „przeciwko chorobie, która ma zostać wyleczona (głównie przewlekłej), należy stosować taką substancję leczniczą, która jest w stanie wywołać inną, najbardziej podobną sztuczną chorobę, a pierwsza zostanie wyleczona”. Similia
similibus (jak jak). Na przykład żółtaczkę należy leczyć żółtym itp.
2) Drugą zasadą jest leczenie bardzo małymi dawkami. Rozcieńczenia leków stosowanych przez homeopatów są liczone w kilku rzędach wielkości, czasami osiągając kilkadziesiąt z nich: 10 w piątym; 10 w dziesiątym; 10 do osiemnastej lub większej potęgi (czyli miliona
i więcej niż ułamek grama). Aby wyjaśnić efekt użycia le
substancje węglowodanowe w
wysokie rozcieńczenia Hahnemann przedstawił spekulatywną koncepcję: „Małe dawki wyróżniają się szczególną siłą duchową, większą aktywnością, zdolnością wnikania do dotkniętych narządów i tkanek”.
Nie wiadomo, co ze szczególną mocą duchową, ale życie naukowe ostatniej dekady przedstawiło bardzo mocne dowody na słuszność twierdzenia Hahnemanna. Tak więc na przykład eksperymenty Francuza Jacquesa Bekveniste'a, przeprowadzone przez niego z rozcieńczeniem substancji 10 do potęgi osiemdziesiątej, wykazały, że cząsteczki wody mają „pamięć” na obecność danej substancji, powodując pewien efekt fizjologiczny . Jeśli ten zauważony w niedalekiej przyszłości fakt znajdzie potwierdzenie, to znaczy, jeśli zostanie ustalone, czy cząsteczki wody są źródłem informacji, niewątpliwie staniemy u podstaw największego odkrycia, które może wyjaśnić terapeutyczną skuteczność leków homeopatycznych.
Następnie rozważ rozdział dotyczący FARMAKOLOGICZNYCH ASPEKTÓW TOKSYCZNEGO DZIAŁANIA NARKOTYKÓW, a mianowicie TOKSYKOLOGIA NARKOTYKÓW. Toksykologia leków jest gałęzią farmakologii, która bada toksyczne działanie tych leków. Jednak teraz lepiej o tym mówić działania niepożądane ludzkie ciało na leki. Fakt ten jest znany od dawna, zgromadzono wiele materiału faktograficznego wskazującego na niepożądane reakcje różne stopnie może wystąpić z prawie wszystkimi lekami.
Istnieje wiele klasyfikacji skutków ubocznych leków i powikłań farmakoterapii, ale żadna nie jest idealna. Jednocześnie, w oparciu o zasadę patogenetyczną, wszystkie niepożądane efekty lub reakcje można podzielić na 2 typy:
1) działania niepożądane związane z
a) przedawkowanie narkotyków
b) zatrucie;
2) reakcje toksyczne związane z właściwościami farmakologicznymi leków.
Przedawkowanie zwykle występuje podczas stosowania dużych dawek leków. Szczególnie często dochodzi do przedawkowania podczas przyjmowania leków o niewielkiej szerokości działania terapeutycznego. Na przykład toksyczność jest trudna do uniknięcia podczas stosowania antybiotyków aminoglikozydowych (streptomycyna, kanamycyna, neomycyna). Leki te powodują zaburzenia przedsionkowe i głuchotę, gdy są leczone w dawkach niewiele wyższych niż terapeutyczne. W przypadku niektórych leków po prostu niemożliwe jest uniknięcie powikłań toksycznych (leki przeciwnowotworowe, cytotoksyczne), które uszkadzają wszystkie szybko dzielące się komórki i
obojętny mózg nie znika, jednocześnie skutecznie wpływając na wzrost komórek nowotworowych.
Ponadto przedawkowanie może wiązać się nie tylko ze stosowaniem dużych dawek, ale również ze zjawiskiem kumulacji (glikozydy nasercowe).
Zatrucie może być przypadkowe lub celowe. Zamierzone zatrucie zwykle ma miejsce w celu samobójczym (samobójstwie). W regionie omskim najczęściej w struktura ogólna zatrucie zajmuje zatrucie płynami kauteryzacyjnymi, na drugim miejscu są zatrucia lekami. Są to przede wszystkim zatrucia tabletkami nasennymi, uspokajającymi, FOS, alkoholem, tlenkiem węgla.
Pomimo różnicy czynników etiologicznych środki pomocy na etapach leczenia są zasadniczo podobne.
Zasady te są następujące:
1) WALCZ Z NIEOSŁONĄ ZATRUCIĄ Z UKŁADU POKARMOWEGO. Jest to najczęściej wymagane w przypadku zatrucia doustnego. Najczęstsze ostre zatrucie jest spowodowane spożyciem substancji. Obowiązkowym i pilnym środkiem w tym zakresie jest płukanie żołądka przez rurkę nawet 10-12 godzin po zatruciu. Jeśli pacjent jest przytomny, wykonuje się płukanie żołądka za pomocą duża liczba woda, a następnie wywołanie wymiotów. Wywoływać wymioty mechanicznie... W stanie nieprzytomności płukanie żołądka przeprowadza się pacjentowi przez sondę. Konieczne jest skierowanie wysiłków na adsorbowanie trucizny w żołądku, do którego stosuje się węgiel aktywowany (1 łyżka stołowa w środku lub 20-30 tabletek jednocześnie, przed i po płukaniu żołądka). Żołądek myje się kilka razy po 3-4 godzinach, aż zostanie całkowicie oczyszczony z substancji.
Wymioty są przeciwwskazane w następujących przypadkach:
ze śpiączką;
W przypadku zatrucia płynami żrącymi;
W przypadku zatrucia naftą, benzyną (możliwość wodorowęglanowego zapalenia płuc z martwicą tkanka płucna itp.).
Jeśli ofiarą jest małe dziecko, lepiej użyć roztwory soli w małych objętościach (100-150 ml).
Najlepiej usunąć truciznę z jelit za pomocą środków przeczyszczających z solą fizjologiczną. Dlatego pod koniec płukania do żołądka można wprowadzić 100-150 ml 30% roztworu siarczanu sodu, a jeszcze lepiej siarczanu magnezu. Środki przeczyszczające z soli fizjologicznej są najsilniejszymi, szybko działającymi środkami przeczyszczającymi w jelitach. Ich działanie jest zgodne z prawami osmozy, dlatego w krótkim czasie zaprzestają działania trucizny.
Dobrze jest podawać zarówno środki ściągające (roztwory garbników, herbata, czeremcha), jak i otulające (mleko, białko jaja, olej roślinny).
W przypadku kontaktu skóry z trucizną konieczne jest dokładne spłukanie skóry, najlepiej wodą z korka. Jeśli substancje toksyczne dostaną się przez płuca, należy powstrzymać ich wdychanie, usuwając poszkodowanego z zatrutej atmosfery.
Przy podskórnym wstrzyknięciu substancji toksycznej jej wchłanianie z miejsca wstrzyknięcia można spowolnić przez wstrzyknięcia roztworu adrenaliny
wokół miejsca wstrzyknięcia, a także ochłodzić ten obszar (lód na skórze w miejscu wstrzyknięcia).
2) Drugą zasadą pomocy w ostrym zatruciu jest wpłynięcie na zassaną truciznę, USUNIĘCIE JEJ Z ORGANIZMU.
W celu jak najszybszego usunięcia toksycznej substancji z organizmu stosuje się przede wszystkim wymuszoną diurezę. Istotą tej metody jest połączenie zwiększonego obciążenia wodą z wprowadzeniem aktywnych, silnych diuretyków. Ciało jest zalane pić dużo wody pacjentowi lub dożylne podawanie różnych roztworów (roztwory zastępcze krwi, glukoza itp.). Spośród diuretyków najczęściej stosuje się FUROSEMIDE (lasix) lub MANNIT. Metodą wymuszonej diurezy niejako „myjemy” tkanki pacjenta, uwalniając je od substancji toksycznych. Ta metoda pozwala jedynie usunąć tylko wolne substancje, które nie są związane z białkami i lipidami krwi. Należy zwrócić uwagę na bilans elektrolitów, który przy stosowaniu Ta metoda może być osłabiony z powodu wydalania znacznej ilości jonów z organizmu.
W ostrej niewydolności sercowo-naczyniowej, ciężkiej dysfunkcji nerek i ryzyku rozwoju obrzęku mózgu lub płuc wymuszona diureza jest przeciwwskazana.
Oprócz wymuszonej diurezy stosuje się hemodializę i dializę otrzewnową, gdy krew (hemodializa lub sztuczna nerka) przechodzi przez półprzepuszczalną błonę, uwalniając się od substancji toksycznych lub „przemywanie” jamy otrzewnej roztworem elektrolitu .
METODY DETOKSYKACJI ZEWNĘTRZNEJ. Skuteczną metodą detoksykacji, która stała się powszechna, jest metoda HEMOSORPCJI (limfosorpcji). W tym przypadku substancje toksyczne we krwi są adsorbowane na specjalnych sorbentach (węgiel ziarnisty pokryty białkami krwi, allo śledziona). Ta metoda pozwala skutecznie odtruć organizm w przypadku zatrucia neuroleptykami, środkami uspokajającymi, FOS itp. Metoda hemosorpcji usuwa substancje słabo usuwane przez hemodializę i dializę otrzewnową.
SUBSTYTUCJA KRWI jest stosowana, gdy krwawienie jest połączone z transfuzją krwi dawcy.
3) Trzecią zasadą walki z ostrymi zatruciami jest ROZŁADOWANIE WYSYSANYCH ZATRUĆ poprzez wprowadzenie ANTAGONISTÓW i ANTYDOTÓW.
Antagoniści są szeroko wykorzystywani w ostrych zatruciach. Na przykład atropina w przypadku zatrucia antycholinoesterazą, FOS; nalorfina - w przypadku zatrucia morfiną itp. Zazwyczaj farmakologiczni antagoniści oddziałują konkurencyjnie z tymi samymi receptorami, co substancje, które spowodowały zatrucie. Pod tym względem bardzo ciekawie wygląda tworzenie SPECYFICZNYCH PRZECIWCIAŁ (monoklonalnych) w stosunku do substancji, które są szczególnie często przyczyną ostrego zatrucia (przeciwciała monoklonalne przeciwko glikozydom nasercowym).
W specyficznym leczeniu pacjentów z zatruciem chemicznym skuteczna jest TERAPIA ANTYDOTOWA. ANTYDOTY to środki stosowane do specyficznego wiązania trucizny, neutralnej
lizanie, unieczynnianie trucizn poprzez oddziaływanie chemiczne lub fizyczne.
Tak więc w przypadku zatrucia metalami ciężkimi stosuje się związki, które tworzą z nimi nietoksyczne kompleksy (na przykład unitiol do zatrucia arszenikiem, D-penicylamina, desferal do zatrucia preparatami żelaza itp.).
4) Czwartą zasadą jest przeprowadzenie TERAPII OBJAWOWEJ. Szczególnie bardzo ważne leczenie objawowe uzyskuje się w przypadku zatrucia substancjami, które nie mają specjalnych odtrutek.
Terapia objawowa wspomaga funkcje życiowe: KRĄŻENIE i ODDECH. Stosują glikozydy nasercowe, wazotoniki, środki poprawiające mikrokrążenie, tlenoterapię oraz stymulatory oddechowe. Drgawki są eliminowane przez zastrzyki sibazonu. W przypadku obrzęku mózgu wykonuje się terapię odwodnienia (furosemid, mannitol). stosuj środki przeciwbólowe, popraw stan kwasowo-zasadowy krwi. Kiedy oddech ustaje, pacjent jest przenoszony do: sztuczna wentylacja płuca z kompleksem środków resuscytacyjnych.
Następnie skupimy się na DRUGIM RODZAJU REAKCJI NIEPOŻĄDANYCH, czyli działaniach niepożądanych związanych z właściwościami farmakologicznymi leków. Skutki uboczne leków ujawniają się u 10-20% pacjentów ambulatoryjnych, a 0,5-5% pacjentów wymaga hospitalizacji w celu skorygowania zaburzeń lekowych. Te niepożądane z punktu widzenia patogenezy reakcje mogą być: a) BEZPOŚREDNIE ib) związane ze ZMIENNĄ WRAŻLIWOŚCIĄ organizmu pacjenta.
Zbadajmy BEZPOŚREDNIE REAKCJE TOKSYCZNE. Nazywa się je bezpośrednimi, ponieważ narkotyki są bezpośrednio, bezpośrednio toksyczne system funkcjonalny... Na przykład antybiotyki z serii aminoglikozydów (streptomycyna, kanamycyna, gentamycyna) wykazują NEUROTOKSYCZNOŚĆ, wywierając toksyczny wpływ na narząd słuchu (ototoksyczność) i aparat przedsionkowy. oprócz ta klasa antybiotyki mają toksyczność w odniesieniu do reakcji behawioralnych, objawiających się letargiem, apatią, letargiem, sennością.
Leki mogą wywoływać BEZPOŚREDNIE REAKCJE GLATOTOKSYCZNE. Na przykład fluorotan (środek znieczulający) z wielokrotnymi aplikacjami w krótki czas może mieć wyraźny efekt toksyczny, aż do ostrej żółtej dystrofii wątroby.
Bezpośrednie efekty toksyczne można uzyskać dzięki NEFROTOKSYCZNOŚCI. Ten efekt mają antybiotyki mycynowe - aminoglikozydy. Przepisując leki z tej serii, pacjent wymaga stałego monitorowania stanu badań moczu (białko, krew w moczu itp.).
Kolejnym bezpośrednim efektem toksycznym jest ULTSEROGENICZNY (wrzodziejący). Na przykład wyznaczenie salicylanów, glikokortykoidów, środka przeciwnadciśnieniowego rezerpiny - prowadzi do owrzodzenia błony śluzowej żołądka, co należy wziąć pod uwagę przy przepisywaniu tych klas leków, zwłaszcza u pacjentów już cierpiących na chorobę wrzodową.
Bezpośrednie efekty toksyczne można wyrazić jako EMBRYOTOKSYCZNOŚĆ. Przypomnę, że embriotoksyczny nazywa się niekorzystnym
wpływ leków, niezwiązany z naruszeniem organogenezy, który występuje przed 12 tygodniem ciąży. A toksyczne działanie leków w późniejszym okresie ciąży nazywa się FETOTOKSYKĄ. Należy pamiętać o tym efekcie przepisując leki kobietom w ciąży, prowadząc dla nich farmakoterapię tylko według ścisłych wskazań.
Przykłady: 1) wyznaczenie streptomycyny kobietom w ciąży może prowadzić do głuchoty u płodu (uszkodzenie VIII pary nerwów czaszkowych); 2) tetracykliny niekorzystnie wpływają na rozwój kości płodu; 3) u matki cierpiącej na morfinizm noworodek może być również uzależniony fizycznie od morfiny.
Leki mogą mieć TERATOGENICZNOŚĆ, czyli taki szkodliwy wpływ na różnicowanie tkanek i komórek, co prowadzi do narodzin dzieci z różnymi anomaliami. Na przykład stosowanie TALIDOMIDU jako środka uspokajającego i nasennego, który ma wyraźne działanie teratogenne, doprowadziło do narodzin w krajach Zachodnia Europa kilka tysięcy dzieci z różnymi deformacjami (fokomelia - kończyny podobne do płetw; amelia - brak kończyn; naczyniaki twarzy, anomalie żołądkowo-jelitowe).
Aby zbadać teratogenne działanie substancji, badany jest wpływ leków na zwierzęta, chociaż nie ma bezpośredniej korelacji z wpływem leków na zwierzęta i ludzi. Na przykład w tym samym talidomidzie wykryto teratogenność w eksperymencie na myszach w dawce 250-500 mg / kg masy ciała, podczas gdy u ludzi wynosiła 1-2 mg / kg.
Najbardziej niebezpieczny pod względem działania teratogennego jest I trymestr (szczególnie okres 3-8 tygodnia ciąży), czyli okres organogenezy. W tych okresach szczególnie łatwo jest wywołać poważną anomalię w rozwoju zarodka.
Tworząc nowe leki, należy również mieć na uwadze możliwość wystąpienia tak poważnych negatywnych skutków jak MUTAGENNOŚĆ CHEMICZNA i RAKOTWÓRCZOŚĆ. MUTAGENNOŚĆ to zdolność substancji do wywoływania trwałego uszkodzenia komórki zarodkowej, a zwłaszcza jej aparatu genetycznego, co objawia się zmianą genotypu potomstwa. RAKOTWÓRCZOŚĆ to zdolność substancji do wywoływania rozwoju nowotwory złośliwe... Estrogeny przyczyniają się do rozwoju raka piersi u kobiet w wieku rozrodczym.
Efekty mutagenne i teratogenne mogą pojawić się miesiące, a nawet lata później, co utrudnia określenie ich rzeczywistej aktywności. Teratogenność tkwi w lekach przeciwnowotworowych, kortykosteroidach, androgenach, alkoholu. Cyklofosfamid i niektóre środki hormonalne mają działanie rakotwórcze.
Działania niepożądane podczas używania narkotyków mogą być wyrażone przez rozwój UZALEŻNIENIA OD NARKOTYKÓW lub, bardziej globalnie, przez UZALEŻNIENIE OD NARKOTYKÓW. Istnieje kilka głównych oznak uzależnienia.
1) Jest to obecność ZALEŻNOŚCI PSYCHICZNEJ, czyli takiego stanu, w którym u pacjenta rozwija się nieodparty pociąg psychiczny do wielokrotnego podawania substancji leczniczej, na przykład,
lek.
2) ZALEŻNOŚĆ FIZYCZNA – termin ten oznacza obecność u pacjenta ciężkiej dolegliwości fizycznej bez wielokrotnego wstrzykiwania leku, w szczególności leku. Wraz z ostrym zaprzestaniem podawania leku, który spowodował uzależnienie od narkotyków, rozwija się zjawisko POZBAWIENIA lub ABSTYNCJI. Pojawia się strach, niepokój, melancholia, bezsenność. Być może pojawia się niepokój ruchowy, agresywność. Wiele funkcji fizjologicznych jest zaburzonych. W ciężkich przypadkach objawy odstawienia mogą być śmiertelne.
3) Rozwój TOLERANCJI, czyli uzależnienia. Inne gatunki
Niepożądanymi skutkami spowodowanymi właściwościami samych leków są zaburzenia związane z przesunięciami w układzie immunobiologicznym pacjenta podczas przyjmowania wysoce aktywnych leków. Na przykład stosowanie antybiotyków o szerokim spektrum działania może objawiać się zmianą normalnej flory bakteryjnej organizmu (jelit), realizowaną przez rozwój nadkażenia, dysbiozy, kandydozy. Najczęściej w te procesy biorą udział płuca i jelita.
Kortykosteroidy i terapia immunosupresyjna osłabiają układ odpornościowy, powodując zwiększone ryzyko rozwoju choroba zakaźna, przede wszystkim o charakterze oportunistycznym (pneumocystoza, wirus cytomegalii itp.).
Ta podgrupa reakcji ma 2 typy:
1) REAKCJE ALERGICZNE;
2) Idiosynkrazja. Należy powiedzieć, że negatywne wpływy
związany z
reakcje chemiczne są bardzo powszechne w praktyce medycznej. Ich częstotliwość cały czas rośnie. Powstają niezależnie od dawki podawanego leku, aw ich powstawaniu zaangażowane są mechanizmy odpornościowe. Reakcje alergiczne mogą mieć 2 typy: NADWRAŻLIWOŚĆ TYPU NATYCHMIASTOWEGO, HNT - związane z tworzeniem przeciwciał klasy IgE i IgG4) oraz typu WOLNEGO (nagromadzenie uwrażliwionych limfocytów T i makrofagów).
Obraz kliniczny jest bardzo zróżnicowany: pokrzywka, wysypki skórne, obrzęk naczynioruchowy, choroba posurowicza, astma oskrzelowa, gorączka, zapalenie wątroby itp. Ale najważniejsza jest możliwość wystąpienia wstrząsu anafilaktycznego. Jeśli rozwój reakcji alergicznych wymaga co najmniej dwukrotnego kontaktu pacjenta z lekiem, to rozwój IDIOSYNKRAZJI - nietolerancji leków podczas początkowego kontaktu z ksenobiotykiem, zawsze wiąże się z jakimś WADEM GENETYCZNYM, zwykle wyrażającym się brakiem lub wyjątkowo niska aktywność enzymu. Na przykład stosowanie leku przeciwmalarycznego prymachiny u osób z genetyczną enzymopatią (niedobór aktu G-6-FDG) powoduje powstawanie chinonu, który ma działanie hemolityczne. W obecności tej fermentopatii niebezpieczne jest przepisywanie leków, które są utleniaczami, ponieważ może to prowadzić do hemolizy
erytrocyty, niedokrwistość hemolityczna lekowa (aspiryna, chloramfenikol, chinidyna, prymachina, furadonina).
KILKA SŁÓW O TWORZENIU NOWYCH LEKÓW, OCENIE LEKÓW I ICH NAZEWNICTWIE. Postęp farmakologii charakteryzuje się ciągłym poszukiwaniem i tworzeniem nowych leków. Tworzenie leków zaczyna się od badań chemików i farmakologów, których twórcza współpraca jest absolutnie niezbędna przy odkrywaniu nowych leków. Jednocześnie poszukiwania nowych środków rozwijają się w kilku kierunkach.
Główną drogą jest synteza CHEMICZNA leków, która może być realizowana w formie syntezy KIEROWANEJ lub mieć sposób EMPIRYCZNY. Jeśli ukierunkowana synteza wiąże się z reprodukcją substancji biogennych (insulina, adrenalina, norepinefryna), tworzeniem antymetabolitów (PABA-sulfonamidów), modyfikacją cząsteczek związków o znanej aktywności biologicznej (zmiana struktury acetylocholiny – gongliobakterie higronium) itp., to ścieżka empiryczna polega albo na losowych znaleziskach, albo na wyszukiwaniu przez przesiewanie, czyli przesiewanie różnych związków chemicznych pod kątem aktywności farmakologicznej.
Jednym z przykładów ustaleń empirycznych może być przypadek obstrukcji efektu hipoglikemicznego przy stosowaniu sulfonamidów, co w konsekwencji doprowadziło do powstania syntetycznych sulfonamidowych perforalnych leków przeciwcukrzycowych (butamid, chlorpropamid).
Inny wariant empirycznego sposobu tworzenia leków, METODA PRZESIEWOWA, również jest bardzo pracochłonna. Jest to jednak nieuniknione, zwłaszcza w przypadku zbadania nowa klasa związki chemiczne, których właściwości na podstawie ich struktury są trudne do przewidzenia (sposób nieefektywny). I tu ogromna rola obecnie trwa komputeryzacja badań naukowych.
Obecnie leki uzyskuje się głównie poprzez celowaną syntezę chemiczną, którą można przeprowadzić a) przez podobieństwo (wprowadzenie dodatkowych łańcuchów, rodników) b) przez komplementarność, czyli korespondencję z dowolnymi receptorami tkanek i narządów.
W arsenale leków, oprócz leków syntetycznych, znaczące miejsce zajmują leki i poszczególne substancje z surowców leczniczych pochodzenia roślinnego lub zwierzęcego, a także z różnych minerałów. Są to przede wszystkim leki galenowe, nowogalenowe, alkaloidy, glikozydy. Tak więc morfina, kodeina, papaweryna są otrzymywane z opium, rezerpina jest otrzymywana z serpentyny serpentynowej, a glikozydy nasercowe - digitoksyna, digoksyna - są otrzymywane z naparstnicy; z wielu gruczołów dokrewnych bydła - hormonów, leków immunoaktywnych (insulina, tarczyca, taktivin itp.).
Niektóre leki są produktami odpadowymi grzybów i mikroorganizmów. Przykładem są antybiotyki. Substancje lecznicze pochodzenia roślinnego, zwierzęcego, mikrobiologicznego, grzybowego często służą jako podstawa ich syntezy, a także późniejszych przemian chemicznych i produkcji leków półsyntetycznych i syntetycznych.
Tempo rozwoju leków nabiera tempa dzięki zastosowaniu
rozwój metod inżynierii genetycznej (insulina itp.).
Nowy lek, po przejściu przez wszystkie te „sita” (badania aktywności farmakologicznej, farmakodynamiki, farmakokinetyki, badania skutków ubocznych, toksyczności itp.) zostaje dopuszczony do badań klinicznych. Wykorzystuje metodę "ślepej kontroli", efekt placebo, metodę podwójnej "ślepej kontroli", kiedy ani lekarz, ani pacjent nie wiedzą, kiedy to placebo jest stosowane. Wie tylko specjalna komisja. Badania kliniczne prowadzone są na ludziach, aw wielu krajach na ochotnikach. Tu oczywiście pojawia się wiele prawnych, deontologicznych, moralnych aspektów problemu, które wymagają jasnego ich opracowania, uregulowania i zatwierdzenia przepisów w tym zakresie.
NOMENKLATURA LEKÓW
Wiele leków, które składają się z jednej substancji czynnej, można nazwać ze względu na ich budowę chemiczną. Jednak ze względu na dużą złożoność ich zapamiętywania i niedogodności ich używania, nazwy chemiczne nie są używane w praktyce medycznej.
Obecnie do oznaczania leków stosuje się 2 rodzaje nazw:
1) NIEOPATENTOWANE międzynarodowe, które są zatwierdzone przez oficjalne organy ds. zdrowia i stosowane w krajowych i międzynarodowych farmakopeach;
2) KOMERCYJNE, czyli marki, które są własnością handlową firm farmaceutycznych. Co więcej, jeden i ten sam lek może mieć wiele nazw. Środek uspokajający diazenam ma nazwy firmowe „seduxen”, „sibazon”, „relanium” itp. Niektóre leki mają ponad 100 nazw (na przykład witamina B12). Zazwyczaj produkt leczniczy ma na opakowaniu zarówno nazwę marki, jak i międzynarodową niezastrzeżoną nazwę.
Preferowane jest przepisywanie leków generycznych, co zmniejsza możliwość: błędy medyczne... Leki te są tańsze niż leki markowe. Ponadto przepisywanie leków pod ich nazwą generyczną umożliwia aptece dostarczenie pacjentowi leku dowolnej firmy produkującej ten lek.
Dla klinicystów najwygodniejszą klasyfikacją leków jest klasyfikacja oparta na ZASADZIE NOZOLOGICZNEJ (na przykład leki stosowane w leczeniu astmy oskrzelowej, zawału mięśnia sercowego, leków przeciwcukrzycowych itp.). Jednak najlepsze klasyfikacje weź pod uwagę takie oznaki leków, jak lokalizacja działania, działanie farmakologiczne, zastosowanie terapeutyczne... Jedną z takich klasyfikacji, która jest najdoskonalsza, jest klasyfikacja akademika M.D. Maszkowskiego, zgodnie z którą przedstawiono również jego znaną książkę informacyjną.
1. Pojęcie leczenia jako ukierunkowanej korekty zaburzeń fizjologicznych organizmu. Korzyści i zagrożenia związane ze stosowaniem leków. Podstawy ich stosowania. Ocena bezpieczeństwa.
Farmakologia- teoretyczne podstawy farmakoterapii.
Powody używania narkotyków:
1) skorygować i wyeliminować przyczynę choroby
2) w przypadku niewystarczających środków zapobiegawczych
3) ze względów zdrowotnych
4) oczywista potrzeba oparta na poziomie wiedzy i doświadczenia
5) dążenie do poprawy jakości życia
Korzyści z przepisywania leków:
1) korekta lub eliminacja przyczyny choroby
2) łagodzenie objawów choroby, jeżeli nie można jej wyleczyć
3) zastąpienie substancji leczniczych naturalnymi substancjami biologicznie czynnymi, niewytwarzanymi przez organizmy w wystarczających ilościach
4) wdrożenie profilaktyki chorób (szczepionki itp.)
Ryzyko- prawdopodobieństwo, że szkoda lub szkoda wyniknie z narażenia; równa się stosunkowi liczby zdarzeń niepożądanych (awersyjnych) do wielkości grupy ryzyka.
A) niedopuszczalne (szkoda > korzyść)
B) akceptowalne (korzyść > szkoda)
B) nieznaczna (105 - poziom bezpieczeństwa)
D) świadomy
Ocena bezpieczeństwa leków zaczyna się na poziomie laboratoria chemiczne syntetyzowanie leków. Przedkliniczna ocena bezpieczeństwa leku jest przeprowadzana przez Ministerstwo Zdrowia, FDA itp. Jeśli lek zostanie pomyślnie zaliczony ten etap rozpoczyna się jego ocena kliniczna, składająca się z czterech faz: I faza – ocena tolerancji u zdrowych ochotników w wieku 20-25 lat, II faza – u chorych ochotników poniżej 100 osób cierpiących na daną chorobę, III faza – wieloośrodkowe badania kliniczne na duże grupy osób (do 1000 osób), IV faza – monitorowanie leku przez 5 lat po jego oficjalnej akceptacji. Jeśli lek pomyślnie przejdzie wszystkie te fazy, jest uważany za bezpieczny.
2. Istota farmakologii jako nauki. Działy i dziedziny współczesnej farmakologii. Główne terminy i koncepcje farmakologii to aktywność farmakologiczna, działanie, skuteczność chemikaliów.
Farmakologia- nauka o lekach we wszystkich aspektach - teoretyczne podstawy terapii:
A) nauka o interakcji chemikaliów z żywymi systemami
B) nauka o zarządzaniu procesami życiowymi organizmu za pomocą chemikaliów.
Działy współczesnej farmakologii:
1) Farmakodynamika- badania a) wpływ leków na organizm człowieka, b) interakcje różnych leków w organizmie podczas ich przepisywania, c) wpływ wieku i różnych chorób na działanie leków
2) Farmakokinetyka- bada wchłanianie, dystrybucję, metabolizm i wydalanie leków (czyli jak organizm pacjenta reaguje na leki)
3) Farmakogenetyka- bada rolę czynników genetycznych w kształtowaniu odpowiedzi farmakologicznej organizmu na leki
4) Farmakoekonomika- ocenia efekty stosowania i koszt leków w celu podjęcia decyzji o ich późniejszym praktycznym zastosowaniu
5) Farmakoepidemiologia- bada stosowanie leków i ich skutki na poziomie populacji lub dużych grup ludzi w celu zapewnienia stosowania najskuteczniejszych i najbezpieczniejszych leków
Aktywność farmakologiczna (biologiczna)- właściwość substancji do wywoływania zmian w biosystemie (ciało ludzkie). Substancje farmakologiczne = substancje biologicznie czynne (BAS)
efekt farmakologiczny- wpływ narkotyków na obiekt i jego cele
Efekt farmakologiczny- wynik działania substancji w organizmie (modyfikacja procesów fizjologicznych, biochemicznych, struktur morfologicznych) - ilościowa, ale nie jakościowa zmiana stanu biosystemów (komórek, tkanek, narządów).
Skuteczność leków- zdolność leków do wywoływania pewnych efektów farmakologicznych niezbędnych w tym przypadku w organizmie. Oceniany na podstawie „istotnych dowodów” – adekwatnych, dobrze kontrolowanych badań i badań klinicznych prowadzonych przez ekspertów z odpowiednim wykształceniem naukowym i doświadczeniem w badaniach leków tego typu (FDA)
3. Chemiczna natura leków. Czynnikami zapewniającymi efekt terapeutyczny leków są działanie farmakologiczne i efekt placebo.
Istnieją leki 1) roślinne 2) zwierzęce 3) mikrobiologiczne 4) mineralne 5) syntetyczne
Leki syntetyczne są reprezentowane przez prawie wszystkie klasy związków chemicznych.
efekt farmakologiczny- wpływ leków na obiekt i jego cel.
Placebo- jakikolwiek składnik terapii, który nie ma żadnego specyficznego biologicznego wpływu na leczoną chorobę.
Jest używany w celu kontroli przy ocenie działania leków oraz w celu uzyskania korzyści dla pacjenta bez żadnych środków farmakologicznych w wyniku jedynie wpływu psychologicznego (tj. Efekt placebo).
Wszystkie rodzaje leczenia mają składnik psychologiczny lub satysfakcjonujący ( Efekt placebo) lub niepokojące ( Efekt nocebo). Przykład efektu placebo: szybka poprawa u pacjenta z infekcją wirusową przy stosowaniu antybiotyków, które nie wpływają na wirusy.
Korzyść z efektu placebo jest związana z psychologicznym wpływem na pacjenta. Będzie maksymalny tylko podczas korzystania z niego. W połączeniu z terapiami które mają wyraźny specyficzny efekt. Drogie substancje jako placebo również pomagają osiągnąć większą odpowiedź.
Wskazania do stosowania placebo:
1) słabe zaburzenia psychiczne
2) wsparcie psychologiczne dla pacjenta z nieuleczalną chorobą przewlekłą lub podejrzanego o trudną diagnozę
4. Źródła i etapy powstawania leków. Definicja pojęć substancji leczniczej, produktu leczniczego, produktu leczniczego i postaci dawkowania. Nazwa leków.
Źródła powstawania leków:
A) surowce naturalne: rośliny, zwierzęta, minerały itp. (glikozydy nasercowe, insulina wieprzowa)
B) modyfikowane naturalne substancje biologicznie czynne
B) związki syntetyczne
D) produkty inżynierii genetycznej (rekombinowana insulina, interferony)
Etapy tworzenia leku:
1. Synteza leków w laboratorium chemicznym
2. Ocena przedkliniczna działania i działań niepożądanych leków Ministerstwa Zdrowia i innych organizmów
3. Badania kliniczne leków (więcej szczegółów w rozdziale 1)
Medycyna- wszelkie substancje lub produkty stosowane do modyfikowania lub badania układów fizjologicznych lub stanów patologicznych z korzyścią dla biorcy (zgodnie z WHO, 1966); poszczególne substancje, mieszaniny substancji lub kompozycje o nieznanym składzie o potwierdzonych właściwościach leczniczych.
Substancja lecznicza- indywidualny związek chemiczny stosowany jako lek.
Forma dawkowania- wygodna forma do praktycznego zastosowania, podana lekowi w celu uzyskania wymaganego efektu terapeutycznego lub profilaktycznego.
Produkt leczniczy- produkt leczniczy w określonej postaci dawkowania, zatwierdzony przez organ rządowy.
5. Sposoby wprowadzania leków do organizmu i ich charakterystyka. Przedsystemowa eliminacja leków.
1. O działanie systemowe
ALE. Droga podania dojelitowego: ustny, podjęzykowy, policzkowy, doodbytniczy, rurkowy
B. Pozajelitowa droga podawania: dożylnie, podskórnie, domięśniowo, wziewnie, podpajęczynówkowo, przezskórnie
2. W przypadku narażenia miejscowego: skórna (naskórkowa), na błonach śluzowych, w jamie brzusznej, opłucnej, stawowej, w tkance (naciek)
|
Droga podania leku |
Zalety |
Wady |
|
Doustnie - doustnie |
1. Wygodny i łatwy dla pacjenta 2. Sterylność leków nie jest wymagana |
1. Wchłanianie wielu leków uzależnione jest od przyjmowanych pokarmów, stanu funkcjonalnego przewodu pokarmowego i innych czynników, które rzadko są brane pod uwagę w praktyce 2. Nie wszystkie leki są dobrze wchłaniane w przewodzie pokarmowym 3. Niektóre leki ulegają zniszczeniu w żołądku (insulina, penicylina) 4. Część leku ma NLR na błonie śluzowej przewodu pokarmowego (NLPZ - objawy śluzówkowe, leki zobojętniające sok żołądkowy - hamują zdolności motoryczne) 5. Nie dotyczy pacjentów w stanie nieprzytomności i z zaburzeniami połykania |
|
podjęzykowe i policzkowe |
1. Wygodne i szybkie wprowadzenie 2. Szybkie wchłanianie leków 3. Leki nie podlegają eliminacji przedukładowej 4. Działanie leku można szybko przerwać |
1. Niedogodności spowodowane częstym regularnym stosowaniem tabletek 2. Podrażnienie błony śluzowej jamy ustnej, nadmierne ślinienie, które przyczynia się do połykania leków i zmniejszenia ich skuteczności 3. Zły smak |
|
Odbytniczo |
1. Połowa leków nie podlega metabolizmowi przedukładowemu 2. Błona śluzowa przewodu pokarmowego nie jest podrażniona 3. Wygodne, gdy inne drogi podania są niedopuszczalne (wymioty, choroba morska, dzidziusie) 4. Działania lokalne |
1. Nieprzyjemne chwile psychologiczne dla pacjenta 2. Wchłanianie leków ulega znacznemu spowolnieniu, gdy odbytnica nie jest opróżniana. |
|
Donaczyniowa (zwykle dożylna |
1. Szybki wstęp do krwi (stany awaryjne) 2. Szybkie tworzenie wysokiej koncentracji systemowej i umiejętność jej zarządzania 3. Umożliwia wprowadzenie leków, które ulegają zniszczeniu w przewodzie pokarmowym |
1. Trudności techniczne dostępu śródnaczyniowego 2. Ryzyko infekcji w miejscu wstrzyknięcia 3. Zakrzepica żył w miejscu wstrzyknięcia leków (erytromycyna) i ból(chlorek potasu) 4. Niektóre leki są adsorbowane na ściankach zakraplaczy (insulina) |
|
domięśniowo |
Wystarczająco szybkie wchłanianie leku do krwi (10-30 min) |
Ryzyko lokalnych powikłań |
|
Podskórnie |
1. Pacjent może samodzielnie wykonywać zastrzyki po treningu. 2. Długoterminowe działanie leków |
1. Powolne wchłanianie i manifestacja działania leku 2. Zanik tkanki tłuszczowej w miejscu wstrzyknięcia i zmniejszenie szybkości wchłaniania leków |
|
Inhalacja |
1. Szybki start akcja i wysokie stężenie w miejscu wstrzyknięcia w leczeniu chorób układu oddechowego. sposoby 2. Dobra sterowność akcji 3. Redukcja toksycznych efektów ogólnoustrojowych |
1. Potrzeba specjalnego urządzenia (inhalatora) 2. Trudności w stosowaniu aerozoli pod ciśnieniem u niektórych pacjentów |
|
Lokalny PM |
1. Wysoka skuteczna koncentracja leków w miejscu wstrzyknięcia 2. Unika się niepożądanych ogólnoustrojowych działań tego leku |
W przypadku naruszenia integralności skóry lek może dostać się do krążenia ogólnoustrojowego - przejaw niepożądanych efektów ogólnoustrojowych. |
Przedukładowa eliminacja leków (efekt pierwszego przejścia)- proces biotransformacji leku przed wejściem leku do krążenia ogólnoustrojowego. Układy enzymatyczne jelita, krew żyły wrotnej i hepatocyty biorą udział w eliminacji przedukładowej po podaniu doustnym leku.
Po podaniu dożylnym nie ma eliminacji przedukładowej.
Aby lek podawany doustnie miał korzystne działanie, konieczne jest zwiększenie jego dawki, aby zrekompensować straty.
6. Transport leków przez bariery biologiczne i jego odmiany. Główne czynniki wpływające na transport leków w organizmie.
Sposoby wchłaniania (transportu) leków przez błony biologiczne:
1) Filtracja (dyfuzja wody) - bierny ruch cząsteczek substancji wzdłuż gradientu stężeń przez pory wypełnione wodą w błonie każdej komórki oraz pomiędzy sąsiednimi komórkami, typowe dla wody, niektórych jonów, małych cząsteczek hydrofilowych (mocznik).
2) Dyfuzja pasywna (dyfuzja lipidów) jest głównym mechanizmem przenoszenia leku, procesu rozpuszczania leku w lipidach błonowych i przemieszczania się przez nie.
3) Transport za pomocą specyficznych nośników - transfer leków za pomocą nośników wbudowanych w błonę (zwykle białek) jest charakterystyczny dla hydrofilowych cząsteczek polarnych, szeregu jonów nieorganicznych, cukrów, aminokwasów, pirymidyn:
a) dyfuzja ułatwiona – prowadzona wzdłuż gradientu stężeń bez zużycia ATP
b) transport aktywny – wbrew gradientowi stężeń z kosztami ATP
Proces nasycenia - to znaczy, że szybkość wchłaniania wzrasta tylko do momentu, gdy liczba cząsteczek leku zrówna się z liczbą nośników.
4) Endocytoza i pinocytoza - lek wiąże się ze specjalnym rozpoznającym składnikiem błony komórkowej, dochodzi do inwazji błony i tworzy się pęcherzyk zawierający cząsteczki leku. Następnie lek jest uwalniany z pęcherzyka do komórki lub transportowany z komórki. Typowe dla polipeptydów o dużej masie cząsteczkowej.
Czynniki wpływające na transport leków w organizmie:
1) właściwości fizyczne i chemiczne substancji (hydro- i lipofilność, jonizacja, polaryzowalność, wielkość cząsteczkowa, stężenie)
2) struktura barier transferowych
3) przepływ krwi
7. Transport przez błony substancji leczniczych o zmiennej jonizacji (równanie jonizacji Hendersona-Hasselbalcha). Zasady kontroli transferu.
Wszystkie leki są słabymi kwasami lub słabymi zasadami, które mają własne wartości stałej jonizacji (pK). Jeżeli wartość pH podłoża jest równa wartości pK leku, to 50% jego cząsteczek będzie w stanie zjonizowanym, a 50% w stanie niezjonizowanym, a podłoże dla leku będzie obojętne.
W środowisku kwaśnym (pH poniżej pK), gdzie występuje nadmiar protonów, słaby kwas będzie w postaci niezdysocjowanej (R-COOH), czyli będzie związany z protonem - protonowany. Ta forma kwasu jest nienaładowana i łatwo rozpuszczalna w lipidach. Jeśli pH zostanie przesunięte na stronę zasadową (tj. pH stanie się większe niż pK), wtedy kwas zacznie dysocjować i straci proton, przechodząc w formę nieprotonowaną, która ma ładunek i jest słabo rozpuszczalna w lipidach .
W środowisku alkalicznym, gdzie występuje niedobór protonów, słaba zasada będzie w formie niezdysocjowanej (R-NH2), czyli będzie nieprotonowana i pozbawiona ładunku. Ta forma bazy jest dobrze rozpuszczalna w tłuszczach i szybko wchłaniana. W środowisku kwaśnym występuje nadmiar protonów i słaba zasada zacznie się dysocjować, wiążąc protony i tworząc protonowaną, naładowaną formę zasady. Ta forma jest słabo rozpuszczalna w lipidach i słabo wchłaniana.
W konsekwencji, Wchłanianie słabych kwasów odbywa się głównie w środowisku kwaśnym, a słabych zasad w środowisku zasadowym.
Cechy metabolizmu słabych kwasów (SC):
1) żołądek: SA w kwaśnej treści żołądka nie ulega jonizacji, aw zasadowym środowisku jelita cienkiego ulega dysocjacji i cząsteczki SA nabierają ładunku. Dlatego wchłanianie słabych kwasów będzie najintensywniejsze w żołądku.
2) we krwi podłoże jest wystarczająco alkaliczne, a wchłonięte cząsteczki SC przekształcą się w formę zjonizowaną. Filtr kłębuszków nerkowych umożliwia przechodzenie zarówno zjonizowanych, jak i niezjonizowanych cząsteczek, dlatego pomimo ładunku cząsteczki SC zostanie wydalony z moczem pierwotnym
3) jeśli mocz ma odczyn zasadowy, kwas pozostanie w formie zjonizowanej, nie będzie mógł ponownie wchłonąć się do krwiobiegu i zostanie wydalony z moczem; Jeśli mocz jest kwaśny, lek przyjmie postać niezjonizowaną, która jest łatwo wchłaniana z powrotem do krwi.
Cechy metabolizmu słabych zasad: przeciwnie do SC (lepsze wchłanianie w jelicie; w moczu zasadowym są ponownie wchłaniane)
To., Aby przyspieszyć eliminację słabego kwasu z organizmu, mocz musi być alkalizowany, a w celu przyspieszenia eliminacji słabej zasady musi być zakwaszony (detoksykacja wg Popowa).
Ilościowa zależność procesu jonizacji leku przy różnym pH środowiska pozwala na otrzymanie równania Henderson— Hasselbach:
Gdzie pKa odpowiada wartości pH, przy której stężenia postaci zjonizowanej i niezjonizowanej są w równowadze .
Równanie Hendersona-Hasselbacha pozwala oszacować stopień jonizacji leku przy danej wartości pH oraz przewidzieć prawdopodobieństwo jego przeniknięcia przez błonę komórkową.
(1)Dla rozcieńczonego kwasu A,
HA ↔ H + + A -, gdzie HA jest stężeniem niezjonizowanej (protonowanej) formy kwasu, a A - jest stężeniem formy zjonizowanej (nieprotonowanej).
(2) Dla słaba podstawa, B,
BH + ↔ H + + B, gdzie BH + to stężenie protonowanej formy zasady, B to stężenie formy nieprotonowanej
Znając pH pożywki i pKa substancji można wyliczonym logarytmem określić stopień jonizacji leku, a co za tym idzie stopień jego wchłaniania z przewodu pokarmowego, reabsorpcji lub wydalania przez nerki przy różnych wartościach pH moczu itp.
8. Przenoszenie leków w organizmie. Dyfuzja wody i dyfuzja w lipidach (prawo Ficka). Transport aktywny.
Przenoszenie leków w organizmie może odbywać się przez dyfuzję wody i lipidów, transport aktywny, endo - i pinocytozę.
Cechy przenoszenia leków w organizmie przez dyfuzję wody:
1. Powłoki nabłonkowe (błony śluzowe przewodu pokarmowego, jamy ustnej itp.) - dyfuzja wody tylko bardzo małych cząsteczek (metanol, jony litu itp.)
2. Kapilary (z wyjątkiem mózgowych) - filtracja substancji o masie cząsteczkowej do 20-30 tysięcy Tak.
3. Naczynia włosowate mózgu - w zasadzie nie posiadają porów wodnych, z wyjątkiem okolic przysadki mózgowej, szyszynki, komory IV strefy, splotu naczyniówkowego, wzniosłości środkowej
4. Łożysko - nie ma porów wodnych (choć kontrowersyjny problem).
5. Wiązanie leków z białkami krwi zapobiega ich uwalnianiu z krwiobiegu, a tym samym dyfuzji wody
6. Dyfuzja w wodzie zależy od wielkości cząsteczek leku i porów wodnych
Cechy dyfuzji lipidów:
1. Główny mechanizm przenoszenia leku przez błony komórkowe
2. Wyznaczony przez lipofilność substancji dyfundującej (tj. współczynnik dystrybucji „olej/woda”) i gradient stężenia, może być ograniczony bardzo niską rozpuszczalnością substancji w wodzie (co zapobiega przenikaniu leku do faza wodna membran)
3. Związki niepolarne łatwo dyfundują, jony są trudne do dyfuzji.
Każda dyfuzja (zarówno wody, jak i lipidów) podlega prawu dyfuzji Ficka:
Szybkość dyfuzji - liczba cząsteczek leku przenoszonych w jednostce czasu; C1 to stężenie substancji na zewnątrz membrany; C2 to stężenie substancji z wnętrza membrany.
Wniosek z prawa Ficka:
1) filtracja leku jest tym większa, im większe jest jego stężenie w miejscu wstrzyknięcia (S powierzchni wchłoniętej w jelicie jest większe niż w żołądku, dlatego wchłanianie leku do jelita jest szybsze)
2) im wyższe stężenie leku w miejscu wstrzyknięcia, tym wyższa filtracja leku
3) filtracja leków jest tym większa, im mniejsza jest grubość błony biologicznej do pokonania (grubość bariery w pęcherzykach płucnych jest znacznie mniejsza niż w skórze, dlatego szybkość wchłaniania jest wyższa w płuca)
Transport aktywny- transfer leków, niezależnie od gradientu stężeń z wykorzystaniem energii ATP, jest charakterystyczny dla hydrofilowych cząsteczek polarnych, szeregu jonów nieorganicznych, cukrów, aminokwasów, pirymidyn. Cechuje: a) selektywność dla niektórych związków b) możliwość konkurowania dwóch substancji o jeden mechanizm transportu c) nasycenie przy wysokich stężeniach substancji d) możliwość transportu wbrew gradientowi stężeń e) zużycie energii.
9. Centralnym postulatem farmakokinetyki jest stężenie leku we krwi - główny parametr kontroli efektu terapeutycznego. Problemy rozwiązywane na podstawie znajomości tego postulatu.
Centralny postulat (dogmat) farmakokinetyki: stężenie leku w osoczu krwi determinuje (określa ilościowo) działanie farmakologiczne.
W większości przypadków szybkość wchłaniania, dystrybucji, metabolizmu i wydalania leków jest proporcjonalna do ich stężenia w osoczu krwi (przestrzega prawa działania masy), więc wiedząc, że jest to możliwe:
1) określić okres półtrwania (dla leków o kinetyce pierwszego rzędu)
2) wyjaśnić czas trwania niektórych toksycznych efektów leków (dla leków w dużych dawkach o kinetyce nasycenia)
10. Biodostępność leków – definicja, istota, ekspresja ilościowa, determinanty. Biodostępność
Biodostępność (F) - charakteryzuje kompletność i szybkość wchłaniania leków przy pozaustrojowych drogach podania - odzwierciedla ilość niezmienionej substancji, która dotarła do krążenia ogólnoustrojowego, w stosunku do początkowej dawki leku.
F wynosi 100% dla leków podawanych dożylnie. Przy podawaniu innymi drogami F jest zwykle mniejsze z powodu niepełnego wchłaniania i częściowego metabolizmu w tkankach obwodowych. F wynosi 0, jeśli lek nie jest wchłaniany ze światła przewodu pokarmowego.
Aby oszacować F, wykreśla się krzywą jako funkcję stężenia leku we krwi w funkcji czasu po jego podaniu dożylnym, jak również po podaniu badaną drogą. To jest tzw. krzywe farmakokinetyczne zależności „czas-stężenie”. Poprzez całkowanie znajduje się wartości obszaru pod krzywą farmakokinetyczną i F oblicza się jako stosunek:
![]() ≤ 1, gdzie AUC jest polem pod krzywą
≤ 1, gdzie AUC jest polem pod krzywą
Biodostępność >70% uważana jest za wysoką, poniżej 30% za niską.
Determinanty biodostępności:
1) prędkość ssania
2) kompletność wchłaniania - niedostateczne wchłanianie leków ze względu na bardzo wysoką hydrofilowość lub lipofilność, metabolizm przez bakterie jelitowe przy podawaniu dojelitowym itp.
3) eliminacja przedukładowa - przy wysokiej biotransformacji w wątrobie leki F są niskie (nitrogliceryna przy podawaniu doustnym).
4) postać dawkowania - tabletki podjęzykowe i czopki doodbytnicze pomóc lekom uniknąć przedukładowej eliminacji.
11. Dystrybucja leków w organizmie. Przedziały, ligandy. Główne determinanty dystrybucji.
Dystrybucja Narkotyki - proces rozprzestrzeniania się leków przez narządy i tkanki po wejściu do krążenia ogólnoustrojowego.
Pola dystrybucyjne:
1. Przestrzeń zewnątrzkomórkowa (osocze, płyn międzykomórkowy)
2. Komórki (cytoplazma, błona organelli)
3. Tkanka tłuszczowa i kostna (odkładanie leków)
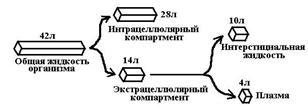 U osoby ważącej 70 kg objętość płynnych mediów wynosi łącznie 42 litry, to jeśli:
U osoby ważącej 70 kg objętość płynnych mediów wynosi łącznie 42 litry, to jeśli:
[Vd = 3-4 L, wtedy cały lek jest rozprowadzany we krwi;
[Vd = 4-14 L, wtedy cały lek jest rozprowadzany w płynie pozakomórkowym;
[Vd = 14-42 l, wtedy cały lek jest w przybliżeniu równomiernie rozprowadzany w organizmie;
[Vd> 42 L, to cały lek znajduje się głównie w przestrzeni pozakomórkowej.
Molekularne ligandy leków:
A) receptory specyficzne i niespecyficzne
B) białka krwi (albumina, glikoproteina) i tkanek
C) polisacharydy tkanki łącznej
D) nukleoproteiny (DNA, RNA)
Determinanty dystrybucji:
· Charakter narkotyków- Jak mniejsze rozmiary molekuły i leki lipofilne, tym szybciej i bardziej równomierne jego rozmieszczenie.
· Rozmiar narządu- Jak większy rozmiar narząd, tym bardziej lek może do niego wejść bez znaczącej zmiany gradientu stężenia
· Przepływ krwi przez organy- w tkankach dobrze ukrwionych (mózg, serce, nerki) stężenie terapeutyczne substancji powstaje znacznie wcześniej niż w tkankach słabo ukrwionych (tłuszcz, kość)
· Obecność barier histohematogennych- leki łatwo przenikają do tkanek ze słabo wyrażonym GHB
· Wiązanie białek osocza- im większa jest frakcja związanego leku, tym gorsza jest jego dystrybucja w tkance, ponieważ tylko wolne cząsteczki mogą opuścić naczynia włosowate.
· Osadzanie leku w tkankach- wiązanie leków z białkami tkankowymi przyczynia się do ich akumulacji w nich, ponieważ zmniejsza się stężenie wolnych leków w przestrzeni okołonaczyniowej i stale utrzymuje się wysoki gradient stężenia między krwią a tkankami.
Ilościową cechą dystrybucji leku jest pozorna objętość dystrybucji (Vd).
Pozorna objętość dystrybucjiVd Jest hipotetyczną objętością płynu, w której można rozłożyć całą podaną dawkę leku w celu uzyskania stężenia równego stężeniu w osoczu krwi.
Vd jest równe stosunkowi podanej dawki (całkowita ilość leku w organizmie) do jego stężenia w osoczu krwi:
 .
.
Im większa pozorna objętość dystrybucji, tym więcej leków jest rozprowadzanych w tkance.
12. Stała eliminacji, jej istota, wymiar, związek z innymi parametrami farmakokinetycznymi.
Stała szybkości eliminacji(kel, min-1) - pokazuje, jaka część leków jest wydalana z organizmu w jednostce czasu Þ Kel = Avid / Atot, gdzie Avid to ilość leków uwalnianych w jednostkach. czas, Absh - całkowita ilość leków w ciele.
Wartość kel zwykle wyznacza się rozwiązując równanie farmakokinetyczne opisujące proces eliminacji leku z krwi, dlatego kel jest nazywany modelowym indeksem kinetycznym. Kel nie jest bezpośrednio związany z planowaniem schematu dawkowania, ale jego wartość jest wykorzystywana do obliczania innych parametrów farmakokinetycznych.
Stała eliminacji jest wprost proporcjonalna do klirensu i odwrotnie proporcjonalna do objętości dystrybucji (z definicji klirensu): Kel = CL / Vd; = godzina-1 / min-1 = ułamek na godzinę.
13. Okres półtrwania leków, jego istota, wymiar, związek z innymi parametrami farmakokinetycznymi.
Okres półeliminacji(t½, min) to czas potrzebny do zmniejszenia stężenia leków we krwi dokładnie o połowę. W tym przypadku nie ma znaczenia, w jaki sposób osiąga się zmniejszenie stężenia - za pomocą biotransformacji, wydalania lub połączenia obu procesów.
Okres półtrwania określa wzór:
![]()
Okres półtrwania jest najważniejszym parametrem farmakokinetycznym, który umożliwia:
B) określić czas całkowitego wyeliminowania leku
C) przewidzieć stężenie leków w dowolnym momencie (dla leków o kinetyce pierwszego rzędu)
14. Klirens jako główny parametr farmakokinetyczny do zarządzania schematem dawkowania. Jego istota, wymiar i związek z innymi parametrami farmakokinetycznymi.
Luz(Cl, ml / min) - objętość krwi, która jest usuwana z leków na jednostkę czasu.
Ponieważ osocze (krew) jest „widoczną” częścią objętości dystrybucji, klirens to ułamek objętości dystrybucji, z którego lek jest uwalniany w jednostce czasu. Jeśli oznaczymy całkowitą ilość leku w organizmie przez Ogólny, a kwota, która została przydzielona po Avyd, następnie:
![]() Z drugiej strony z definicji objętości dystrybucji wynika, że całkowita ilość leku w organizmie wynosi Absz =Vd´
CTer / plazma... Podstawiając tę wartość do wzoru na klirens, otrzymujemy:
Z drugiej strony z definicji objętości dystrybucji wynika, że całkowita ilość leku w organizmie wynosi Absz =Vd´
CTer / plazma... Podstawiając tę wartość do wzoru na klirens, otrzymujemy:
![]() .
.
Zatem klirens jest stosunkiem szybkości eliminacji leku do jego stężenia w osoczu krwi.
W tej formie formuła klirensu służy do obliczenia dawki podtrzymującej leku ( DNS), czyli dawka leku, która powinna zrekompensować utratę leku i utrzymać jego poziom na stałym poziomie:
Szybkość wtrysku = Szybkość wydalania =Cl´ CTer(dawka / min)
DNS= szybkość wtrysku´ T (T- przerwa między przyjmowaniem leku)
Prześwit jest addytywny, czyli eliminacja substancji z organizmu może nastąpić przy udziale procesów zachodzących w nerkach, płucach, wątrobie i innych narządach: Clsystem = Clrenal. + Cl wątroba + Cld.
Odprawa ograniczona Z okresem półtrwania leku i objętością dystrybucji: t1 / 2 = 0,7 * Vd / Cl.
15. Dawka. Rodzaje dawek. Jednostki dawkowania leków. Cele dawkowania leków, metody i opcje podawania, odstępy między podawaniem.
Wpływ leków na organizm w dużej mierze zależy od ich dawki.
Dawka- ilość substancji wprowadzonej do organizmu jednorazowo; wyrażone w jednostkach masy, objętości lub konwencjonalnych (biologicznych).
Rodzaje dawek:
A) pojedyncza dawka - ilość substancji na dawkę
B) dawka dzienna - ilość przepisanego leku na dzień w jednej lub kilku dawkach
C) dawka oczywiście - całkowita ilość leku na przebieg leczenia
D) dawki terapeutyczne - dawki, w których lek jest stosowany w celach terapeutycznych lub profilaktycznych (progowe lub minimalne skuteczne, średnie dawki terapeutyczne i wyższe terapeutyczne).
E) dawki toksyczne i śmiertelne - dawki leków, przy których zaczynają wykazywać wyraźne działanie toksyczne lub powodują śmierć organizmu.
E) dawka nasycająca (wstępna) - liczba wstrzykniętych leków, która wypełnia całą objętość dystrybucji organizmu w stężeniu skutecznym (leczniczym): VD = (Css * Vd) / F
G) dawka podtrzymująca - systematycznie podawana ilość leków, która kompensuje utratę leków klirensem: PD = (Css * Cl * DT) / F
Farmaceutyczne jednostki dawkowania:
1) w gramach lub ułamkach grama narkotyków
2) liczba leków na 1 Kg masa ciała (na przykład 1 Mg/kg) lub na jednostkę powierzchni ciała (na przykład 1 Mg/m2)
Cele dawkowania leków:
1) określić ilość leków potrzebną do wywołania pożądanego efektu terapeutycznego przez określony czas
2) unikać zjawisk zatrucia i skutków ubocznych przy wprowadzaniu leków
Metody podawania leku: 1) dojelitowo 2) pozajelitowo (patrz punkt 5)
Opcje podawania leków:
A) ciągły (przez długotrwałą donaczyniową infuzję leków przez kroplówkę lub automatyczne dozowniki). Przy ciągłym podawaniu leków jego stężenie w organizmie zmienia się płynnie i nie ulega znacznym wahaniom.
B) podawanie przerywane (metodą iniekcji lub bez iniekcji) - podawanie leku w regularnych odstępach czasu (odstępy dawkowania). Przy okresowym podawaniu leków jego stężenie w organizmie stale się zmienia. Po przyjęciu określonej dawki najpierw wzrasta, a następnie stopniowo spada, osiągając wartości minimalne przed kolejnym podaniem leku. Wahania stężenia są tym większe, im większa jest podana dawka leku i odstęp między iniekcjami.
Interwał wprowadzenia- odstęp pomiędzy podawanymi dawkami, zapewniający utrzymanie terapeutycznego stężenia substancji we krwi.
16. Podawanie leków w stałym tempie. Kinetyka stężenia leku we krwi. Stacjonarne stężenie leku we krwi ( CSS), czas jego osiągnięcia, obliczenia i zarządzania nim.
 Osobliwością wprowadzania leków w stałym tempie jest płynna zmiana ich stężenia we krwi po podaniu, przy czym:
Osobliwością wprowadzania leków w stałym tempie jest płynna zmiana ich stężenia we krwi po podaniu, przy czym:
1) czas do osiągnięcia stężenia leku w stanie stacjonarnym wynosi 4-5t½ i nie zależy od szybkości wlewu (wielkości podanej dawki)
2) wraz ze wzrostem szybkości wlewu (dawka wstrzyknięta) wartość СSS również wzrasta proporcjonalną liczbę razy
3) eliminacja leku z organizmu po zakończeniu wlewu trwa 4-5t½.
ZSS- równowagowe stężenie stacjonarne- stężenie leków osiągane przy szybkości podawania równej szybkości wydalania, a więc:
![]() (z definicji odprawy)
(z definicji odprawy)
Dla każdego kolejnego okresu półtrwania stężenie leku wzrasta o połowę pozostałego stężenia. Wszystkie leki, które są zgodne z prawem eliminacji pierwszego rzędu, są DosięgnieCSSpo 4-5 okresach półtrwania.
Podejścia do zarządzania na poziomie CSS: zmień podawaną dawkę leków lub przerwę w podawaniu
17. Przerywane podawanie leków. Kinetyka stężenia leków we krwi, zakres stężeń terapeutycznych i toksycznych. Obliczanie stężenia stacjonarnego ( CSS), granice jego oscylacji i jego kontroli. Odpowiednia dyskretna przerwa w dozowaniu.
 Wahania stężenia leków w osoczu krwi: 1 - ze stałą kroplówką dożylną; 2 - z ułamkowym wprowadzeniem tej samej dziennej dawki w odstępie 8 godzin, 3 - z wprowadzeniem dziennej dawki w odstępie 24 godzin.
Wahania stężenia leków w osoczu krwi: 1 - ze stałą kroplówką dożylną; 2 - z ułamkowym wprowadzeniem tej samej dziennej dawki w odstępie 8 godzin, 3 - z wprowadzeniem dziennej dawki w odstępie 24 godzin.
Okresowe podawanie leku- wprowadzanie pewnej ilości leków w odstępach czasu.
Równowagowe stężenie w stanie stacjonarnym osiąga się po 4-5 okresach połowicznej eliminacji, czas do jego osiągnięcia nie zależy od dawki (na początku, gdy poziom stężenia leku jest niski, szybkość jego eliminacji jest również niska; jak zwiększa się ilość substancji w organizmie, wzrasta również tempo jej eliminacji, dlatego wcześnie lub późno przyjdzie moment, w którym zwiększone tempo eliminacji zrównoważy podaną dawkę leku i dalszy wzrost stężenia zatrzyma się)
Css jest wprost proporcjonalna do dawki leku i odwrotnie proporcjonalna do przerwy w podawaniu i klirensu leku.
Granice huśtawki CSS: ![]() ; Cssmin = Cssmax × (1 - e-mail). Wahania stężenia leku są proporcjonalne do T / t1 / 2.
; Cssmin = Cssmax × (1 - e-mail). Wahania stężenia leku są proporcjonalne do T / t1 / 2.
Zasięg terapeutyczny (korytarz bezpieczeństwa, okno terapeutyczne)- Jest to zakres stężeń od minimalnego terapeutycznego do wywołującego pierwsze oznaki skutków ubocznych.
Zasięg toksyczny- zakres stężeń od najwyższych terapeutycznych do śmiertelnych.
Odpowiednie podawanie dyskretnych dawek: sposób podawania, w którym wahania stężenia leku we krwi mieszczą się w zakresie terapeutycznym. Aby określić odpowiedni schemat podawania leku, konieczne jest obliczenie. Różnica między Cssmax i Cssmin nie powinna przekraczać 2Css.
Kontrola oscylacjiCSS:
Zakres huśtawkiCSSwprost proporcjonalna do dawki leków i odwrotnie proporcjonalna do przerwy w jej podawaniu.
1. Zmień dawkę leków: wraz ze wzrostem dawki leku zakres wahań jego Css proporcjonalnie wzrasta
2. Zmień interwał podawania leku: wraz ze wzrostem przerwy w podawaniu leku zakres wahań jego Css proporcjonalnie maleje
3. Jednoczesna zmiana dawki i przerwy w podawaniu
18. Dawka początkowa (ładująca). Znaczenie terapeutyczne, obliczanie parametrów farmakokinetycznych, warunki i ograniczenia jego stosowania.
Dawka początkowa (ładująca)- dawka podana jednorazowo i wypełnia całą objętość dystrybucji w aktualnym stężeniu terapeutycznym. VD = (Css * Vd) / F; = mg / l, = l / kg
Znaczenie terapeutyczne: dawka początkowa szybko zapewnia skuteczne terapeutyczne stężenie leków we krwi, co umożliwia np. szybkie powstrzymanie ataku astmy, arytmii itp.
Dawkę początkową można podać jednorazowo tylko wtedy, gdy Proces dystrybucji substancji jest ignorowany
Ograniczenie korzystania z VD: jeśli lek jest dystrybuowany Znacznie wolniej niż jego wejście do krwioobiegu wprowadzenie od razu całej dawki nasycającej (zwłaszcza dożylnie) spowoduje powstanie stężenia znacznie wyższego od terapeutycznego i spowoduje wystąpienie efektów toksycznych. Warunki użytkowania VD: zatem wprowadzenie dawek obciążeniowych Powinien być zawsze powolny lub ułamkowy.
19. Dawki podtrzymujące, ich znaczenie terapeutyczne i obliczanie optymalny reżim dawkowanie.
Dawka podtrzymująca- dawka leków podawanych systematycznie, która wypełnia objętość klirensu, czyli fragment Vd, który jest usuwany z leków w okresie DT: PD = (Css * Cl * DT) / F.
Znaczenie terapeutyczne: PD kompensuje straty z klirensem w okresie między wstrzyknięciami leku.
Obliczanie optymalnej dawki leków (w celu szybkiego złagodzenia ataku):
1. Oblicz VD: VD = (Css * Vd) / F
2. Wybierz przedział dla wprowadzenia DT (zwykle większość leków jest przepisywana z przedziałem bliskim t1/2) i oblicz PD: PD = (Css * Cl * DT) / F
3. Sprawdzamy, czy wahania leków we krwi nie wykraczają poza zakres terapeutyczny, obliczając Cssmax i Cssmin: ![]() ; Cssmin = Cssmax × (1 - e-mail). Różnica między Cssmax i Cssmin nie powinna przekraczać dwóch Css.
; Cssmin = Cssmax × (1 - e-mail). Różnica między Cssmax i Cssmin nie powinna przekraczać dwóch Css.
Ułamek do wyeliminowania znajduje się na podstawie wykresu (patrz rozdział 16) lub według wzoru: ![]()
4. Jeżeli w wybranym przez nas odstępie podawania leku jego wahania wykraczają poza zakres terapeutyczny, zmień DT i powtórz obliczenia (pkt 2 - pkt 4)
Uwaga! Jeśli lek nie jest przeznaczony do łagodzenia stanów nagłych lub jest przyjmowany w tabletkach, VD nie jest obliczana.
20. Różnice osobnicze, wiekowe i płciowe w farmakokinetyce leków. Korekty obliczania poszczególnych wartości objętości dystrybucji leków.
1. Różnice wieku farmakokinetyka leków.
|
1. Warstwa rogowa naskórka jest cieńsza, dlatego po nałożeniu na skórę leki są lepiej wchłaniane. Wchłanianie leków po podaniu doodbytniczym jest również lepsze. 2. Objętość płynu w organizmie dzieci wynosi 70-80%, podczas gdy u dorosłych tylko »60%, dlatego Vd leków hydrofilowych, które mają, jest wyższe i wymagane są wyższe dawki. 3. U noworodka poziom albuminy w osoczu jest niższy niż u dorosłych, dlatego wiązanie leków z białkiem jest mniej intensywne. 4. Noworodki mają niską intensywność układów cytochromu P450 i enzymów sprzęgających, ale wysoką aktywność układów metylujących. 5. Szybkość filtracji kłębuszkowej w nerkach dzieci w wieku poniżej 6 miesięcy wynosi 30-40% wskaźnika dorosłych, dlatego zmniejsza się wydalanie leków przez nerki. |
1. Występuje spadek stężenia albuminy w osoczu krwi i frakcji leku związanej z białkiem 2. Zawartość wody w organizmie spada z 60% do 45%, dlatego zwiększa się akumulacja leków lipofilowych. 3. Szybkość filtracji kłębuszkowej może spaść do 50-60% wskaźnika dojrzałego pacjenta, dlatego wydalanie leków przez nerki jest znacznie ograniczone. |
2. Różnice płci w działaniu leków... Kobiety charakteryzują się niższą masą ciała niż mężczyźni, dlatego wielkość dawek leków dla nich powinna z reguły znajdować się w dolnej granicy zakresu dawek terapeutycznych.
3. Stany patologiczne organizmu i działanie leków
A) choroba wątroby: leki F z powodu zatrzymania metabolizmu pierwszego przejścia, frakcja leków niezwiązanych z powodu braku syntezy albumin, działanie leków jest przedłużone z powodu ich biotransformacji.
B) patologia nerek: eliminacja leków wydalanych przez nerki spowalnia
4. Czynniki genetyczne- niedobór niektórych enzymów metabolizmu leków może przyczynić się do przedłużenia ich działania (pseudocholinoesteraza itp.)
Korekty do obliczania poszczególnych wartości objętości dystrybucji leków:
A) w przypadku otyłości leki lipofobowe są nierozpuszczalne w tkance tłuszczowej należy obliczyć idealna waga według wzrostu (wzór Broki: idealna waga = wzrost (w cm) - 100) i przelicz Vd według idealnej wagi według wzrostu.
B) przy obrzęku należy obliczyć nadmiar wody = nadwaga- idealnie, Vd należy zwiększyć na litr każdego nadmiarowego kilograma wody.
Zależność głównych parametrów farmakokinetycznych od różnych czynników:
1. Wchłanianie leków: w wieku ¯ wchłanianie leku, jego metabolizm w trakcie eliminacji przedukładowej, zmienia się biodostępność leków.
2. Objętość dystrybucji Vd: ¯ z wiekiem i otyłością, z obrzękiem
3. Okres półtrwania: zmiany wraz z wiekiem i otyłością (od spadku Vd)
4. Klirens: określony przez stan funkcjonalny nerek i wątroby
21. Klirens nerkowy leków, mechanizmy, ich cechy ilościowe i jakościowe.
Klirens nerkowy jest miarą objętości osocza krwi, która jest usuwana z substancji leczniczej w jednostce czasu przez nerki: Cl (ml/min) = U × V/P, gdzie U to stężenie leku w ml mocz, V to objętość wydalanego moczu w min, a P = stężenie leku w ml osocza.
Mechanizmy klirensu nerkowego i ich cechy:
1. Filtrowanie: Wyemitowane leki Tylko filtracja(insulina) będzie miała klirens równy GFR (125-130 ml/min)
Określane przez: przepływ krwi przez nerki, niezwiązaną frakcję leku i zdolność filtracji nerek.
Większość leków ma niską masę cząsteczkową i dlatego jest swobodnie filtrowana z osocza w kłębuszkach nerkowych.
2. Aktywna sekrecja: Wyemitowane leki Filtracja i całkowite wydzielanie(kwas paraaminogipurowy), będzie miał klirens równy klirensowi nerkowo-osoczowemu (650 ml/min)
Cewka nerkowa zawiera dwa systemy transportowe które mogą uwalniać leki do ultrafiltratu, jeden na kwasy organiczne i kolejny dla zasady organiczne. Systemy te wymagają energii do aktywnego transportu wbrew gradientowi stężenia; są miejscem rywalizacji o nosiciela jednych leków z innymi.
Określane przez: maksymalne tempo wydzielania, objętość moczu
3. Reabsorpcja: wartości klirensu między 130 a 650 ml/min sugerują, że lek jest Filtrowane, wydalane i częściowo reabsorbowane
Reabsorpcja zachodzi w całym kanale nerkowym i zależy od polarności leku, niepolarne, lipofilowe są reabsorbowane.
Wyznaczane przez: pierwotną wartość pH i jonizację leku
Szereg wskaźników, takich jak: Wiek, wspólne stosowanie kilku leków, choroby znacząco wpływają na klirens nerkowy:
A) niewydolność nerek ® zmniejszenie klirensu leku wysoki poziom Narkotyki we krwi
B) kłębuszkowe zapalenie nerek ® utrata białka surowicy, która była zwykle dostępna i związana z lekami ® wzrost poziomu wolnej frakcji leków w osoczu
22. Czynniki wpływające na klirens nerkowy leków. Zależność klirensu od właściwości fizykochemicznych leków.
Czynniki wpływające na nerkiCl:
A) filtracja kłębuszkowa
B) szybkość przepływu krwi przez nerki
B) maksymalna szybkość wydzielania
D) objętość moczu
E) frakcja niezwiązana we krwi
Zależność klirensu nerkowego od właściwości fizykochemicznych leków:
Ogólne wzory: 1) leki polarne nie ulegają reabsorbcji, leki niepolarne ulegają reabsorbcji 2) leki jonowe ulegają sekrecji, leki niejonowe nie ulegają sekrecji.
I. Niepolarne substancje niejonowe: filtrowane tylko w postaci niezwiązanej, nie wydzielane, reabsorbowane
Klirens nerkowy jest niewielki i zależy od: a) frakcji niezwiązanych leków we krwi b) objętości moczu
II. Polarne substancje niejonowe: filtrowane w postaci niezwiązanej, nie wydzielają, nie wchłaniają się ponownie
Klirens nerkowy jest wysoki, determinowany przez: a) frakcję niezwiązanych leków we krwi b) szybkość filtracji kłębuszkowej
III. Niepolarny zjonizowany w moczu w postaci niejonowej: filtrowany, aktywnie wydzielany, niepolarny reabsorbowany
Klirens nerkowy jest określany przez: a) frakcję niezwiązanych leków we krwi b) frakcję leków zjonizowanych w moczu c) objętość moczu
IV. Polar zjonizowany w moczu w postaci niezjonizowanej: filtrowany, aktywnie wydzielany, nie wchłaniany
Klirens nerkowy określa: a) nerkowy przepływ krwi i szybkość filtracji kłębuszkowej b) maksymalna szybkość wydzielania
23. Klirens leków przez wątrobę, jego uwarunkowania i ograniczenia. Cykl leków jelitowo-wątrobowych.
Mechanizmy klirensu wątrobowego:
1) metabolizm (biotransformacja) przez utlenianie, redukcję, alkilację, hydrolizę, koniugację itp.
Główna strategia metabolizmu ksenobiotyków: substancje niepolarne ® polarne (hydrofilowe) metabolity wydalane z moczem.
2) wydzielanie (wydalanie nieprzekształconych substancji do żółci)
Do żółci transportowane są tylko substancje polarne o masie cząsteczkowej > 250 aktywnych (kwasy organiczne, zasady).
Determinanty klirensu wątrobowego:
A) Szybkość przepływu krwi w wątrobie
B) Maksymalna prędkość wydalanie lub przemiana metaboliczna
B) Km - stała Michaelisa
D) Frakcja niezwiązana z białkami
Ograniczenia klirensu wątrobowego:
1. Jeśli Vmax / Km jest duże → Cl pecs = prędkość przepływu krwi w wątrobie
2. Jeśli Vmax / Km wartości średnie → Cl = suma wszystkich czynników
3. Jeśli Vmax / Km jest małe → piec Cl jest mały, ograniczony
Cykl leków jelitowo-wątrobowych - Szereg leków i produktów ich przemiany w znacznych ilościach jest wydalany z żółcią do jelit, skąd są częściowo wydalane z ekskrementami, a częściowo - Ponownie wchłonięty do krwi, ponownie wchodzi do wątroby i jest wydalany do jelit.
Eliminacja leków przez wątrobę może ulec znacznej zmianie Choroba wątroby, wiek, dieta, genetyka, czas trwania przepisywania leków(na przykład z powodu indukcji enzymów wątrobowych) i innych czynników.
24. Czynniki zmieniające klirens substancji leczniczych.
1. Oddziaływania leków na poziomie: wydzielania nerkowego, przemiany biochemicznej, zjawiska indukcji enzymatycznej
2. Choroba nerek: zaburzenia przepływu krwi, ostre i przewlekłe uszkodzenie nerek, skutki długotrwałej choroby nerek
3. Choroby wątroby: marskość alkoholowa, marskość pierwotna, zapalenie wątroby, wątrobiaki
4. Choroby przewodu pokarmowego i narządów dokrewnych
5. Indywidualna nietolerancja (brak enzymów acetylacji – nietolerancja aspiryny)
25. Korekta terapii lekowej w chorobach wątroby i nerek. Podejścia ogólne. Korekta schematu dawkowania pod kontrolą całkowitego klirensu leku.
1. Zrezygnuj z leków, które nie są konieczne
2. W przypadku choroby nerek należy stosować leki wydalane w wątrobie i odwrotnie.
3. Zmniejsz dawkę lub zwiększ odstęp między wstrzyknięciami
4. Ścisłe monitorowanie skutków ubocznych i toksycznych
5. W przypadku braku efektu farmakologicznego dawkę należy zwiększać powoli i pod kontrolą efektów farmakologicznych i toksycznych.
6. Jeśli to możliwe, określ stężenie substancji w osoczu i indywidualnie skoryguj terapię zgodnie z Cl leku
7. Stosować pośrednią metodę oceny Cl.
Korekta schematu dawkowania pod kontrolą całkowitego klirensu leku:
Dostosowanie dawki : Dind = Dtyp. × Clind. / Cl.typ.
Przy ciągłym dożylnym podawaniu leku: Indywidualna szybkość podawania = Typowa szybkość podawania × Cl ind. / Cl typowy
Przy podawaniu przerywanym: 1) zmienić dawkę 2) zmienić interwał 3) zmienić oba parametry. Na przykład, jeśli klirens zmniejszy się o 50%, można zmniejszyć dawkę o 50% i zachować odstęp lub podwoić odstęp i zachować dawkę. Korzystne jest zmniejszenie dawki i utrzymanie przerwy w podawaniu.
26. Korekta schematu dawkowania pod kontrolą resztkowej czynności nerek.
Klirens kreatyniny- najważniejszy ilościowy wskaźnik czynności nerek, na podstawie którego można dostosować schemat dawkowania
Wiemy:
A) resztkowa czynność nerek, określona na podstawie klirensu kreatyniny u danego pacjenta Clcr/pacjenta
B) całkowity klirens danego leku (CLP/całkowity) oraz udział nerkowego klirensu leku w całkowitym klirensie
C) prawidłowy klirens kreatyniny Clcr / normogram
3) Css i F dla tej sieci LAN (z odniesienia)
Znaleźć: dawka leku dla tego pacjenta
ClPS / norma nerkowa = ClPS / całkowity udział X klirensu nerkowego leku w całkowitym klirensie
CLP / pacjent z nerkami = Clcr / pacjent / Clcr / norma * ClLS / norma nerek
ClPP / wskaźnik pozanerkowy = ClPP / całkowity - ClPP / wskaźnik nerkowy
ClPS/całkowity pacjent = CLPS/pacjent nerkowy + ClPS/norma pozanerkowa
Dawka tego leku wewnątrz z prawidłową czynnością nerek wynosi: norma PD = Css X Cl / F
Dawka tego leku dla naszego pacjenta jest równa: PD pacjenta = PD norma X СlPS / ogólny pacjent / СlPS / ogółem
Odpowiadać: PDbolny
27. Korekta terapii lekowej uszkodzenia wątroby i innych stanów patologicznych.
Choroba wątroby może zmniejszyć klirens i wydłużyć okres półtrwania wielu leków. Jednak w przypadku niektórych leków, które są eliminowane przez wątrobę, parametry te nie zmieniają się w przypadku dysfunkcji wątroby, dlatego Choroba wątroby nie zawsze wpływa na wewnętrzny klirens wątrobowy... Obecnie nie ma wiarygodnego markera, który można wykorzystać do przewidywania klirensu wątrobowego podobnego do klirensu kreatyniny.
W celu skorygowania schematu dawkowania w przypadku choroby nerek zob. paragraf 26 powyżej, ogólne zasady korekcji zob. paragraf 25.
28. Strategia indywidualnej terapii lekowej.
Uznanie ważnej roli stężenia jako łącznika farmakokinetyki i farmakodynamiki przyczynia się do stworzenia strategii stężenia docelowego – optymalizacji dawki u danego pacjenta w oparciu o pomiar stężenia leku. Składa się z następujących etapów:
1. Wybór stężenia docelowego
2. Oblicz Vd i Cl na podstawie typowych wartości i dokonaj korekt ze względu na takie czynniki, jak masa ciała i czynność nerek.
3. Wpisanie dawki nasycającej lub podtrzymującej, obliczonej z uwzględnieniem wartości TC, Vd i Cl.
4. Rejestracja reakcji pacjenta i określenie stężenia leku
5. Weryfikacja Vd i Cl na podstawie wyników pomiarów stężeń.
6. Powtórz kroki 3-6, aby dostosować dawkę podtrzymującą wymaganą dla optymalnej odpowiedzi na lek.
29. Biotransformacja leków, jej znaczenie biologiczne, główne kierunki i wpływ na działanie leków. Główne fazy przemian metabolicznych leków w organizmie.
Biotransformacja leków- przemiany chemiczne leków w organizmie.
Biologiczne znaczenie biotransformacji leków: stworzenie substratu dogodnego do późniejszej utylizacji (jako materiał energetyczny lub plastik) lub przyśpieszenie eliminacji leków z organizmu.
Główny kierunek przemian metabolicznych leków: leki niepolarne → polarne (hydrofilowe) metabolity wydalane z moczem.
Istnieją dwie fazy reakcji metabolicznych leków:
1) Transformacja metaboliczna (reakcje niesyntetyczne, faza 1)- przemiany substancji w wyniku mikrosomalnego i pozamikrosomalnego utleniania, redukcji i hydrolizy
2) koniugacja (reakcje syntetyczne, faza 2)- proces biosyntezy, któremu towarzyszy dodanie do leku lub jego metabolitów szeregu grup chemicznych lub cząsteczek związków endogennych poprzez a) tworzenie glukuronidów b) estry glicerolu c) sulfoestry d) acetylację e) metylację
Wpływ biotransformacji na aktywność farmakologiczną leków:
1) najczęściej metabolity biotransformacji nie wykazują aktywności farmakologicznej lub ich aktywność jest zmniejszona w porównaniu z substancją wyjściową
2) w niektórych przypadkach metabolity mogą zachować aktywność, a nawet przewyższyć aktywność substancji macierzystej (kodeina jest metabolizowana do bardziej aktywnej farmakologicznie morfiny)
3) czasami podczas biotransformacji powstają substancje toksyczne (metabolity izoniazydu, lidokainy)
4) czasami podczas biotransformacji powstają metabolity o przeciwnych właściwościach farmakologicznych (metabolity nieselektywnych agonistów receptorów b2-adrenergicznych mają właściwości blokerów tych receptorów)
5) szereg substancji to proleki, które początkowo nie dają efektów farmakologicznych, ale w toku biotransformacji są przekształcane w substancje biologicznie czynne (nieaktywna L-dopa, przenikając przez BBB, zamienia się w mózgu w aktywną dopaminę, natomiast tam nie występują ogólnoustrojowe działania dopaminy).
30. Kliniczne znaczenie biotransformacji leków. Wpływ płci, wieku, masy ciała, czynniki środowiskowe, palenie, alkohol na biotransformację narkotyków.
Kliniczne znaczenie biotransformacji leków: ponieważ dawka i częstotliwość podawania wymagane do osiągnięcia skutecznego stężenia we krwi i tkankach mogą się różnić u pacjentów ze względu na indywidualne różnice w dystrybucji, szybkości metabolizmu i eliminacji leków, ważne jest uwzględnienie ich w praktyce klinicznej.
Wpływ różnych czynników na biotransformację leków:
ALE) Stan funkcjonalny wątroba: w przypadku jej chorób klirens leków zwykle maleje, a okres półtrwania eliminacji wzrasta.
B) Wpływ czynników środowiskowych: palenie sprzyja indukcji cytochromu P450, w wyniku czego metabolizm leków ulega przyspieszeniu podczas utleniania mikrosomalnego
W) Wegetarianie spowolnienie biotransformacji leków
D) starsi i młodsi pacjenci charakteryzują się: nadwrażliwość na farmakologiczne lub toksyczne działanie leków (u osób starszych i dzieci do 6 miesiąca życia zmniejsza się aktywność utleniania mikrosomalnego)
E) u mężczyzn metabolizm niektórych leków jest szybszy niż u kobiet, ponieważ androgeny stymulują syntezę mikrosomalnych enzymów wątrobowych (etanol)
MI) Wysoka zawartość białka i intensywność ćwicz stres : przyspieszenie metabolizmu leków.
F) Alkohol a otyłość spowolnić metabolizm leków
31. Metaboliczne interakcje leków. Choroby wpływające na ich biotransformację.
Interakcja metaboliczna leków:
1) indukcja enzymów metabolizmu leków - bezwzględny wzrost ich liczby i aktywności w wyniku ekspozycji na niektóre leki. Indukcja prowadzi do przyspieszenia metabolizmu leków i (z reguły, ale nie zawsze) do zmniejszenia ich aktywności farmakologicznej (ryfampicyna, barbiturany - induktory cytochromu P450)
2) hamowanie enzymów metabolizmu leków - hamowanie aktywności enzymów metabolicznych pod wpływem niektórych ksenobiotyków:
A) konkurencyjne interakcje metaboliczne – leki o dużym powinowactwie do niektórych enzymów zmniejszają metabolizm leków o mniejszym powinowactwie do tych enzymów (werapamil)
B) wiązanie z genem indukującym syntezę niektórych izoenzymów cytochromu P450 (cymedyny)
C) bezpośrednia inaktywacja izoenzymów cytochromu P450 (flawonoidy)
Choroby wpływające na metabolizm leków:
A) choroba nerek (zaburzenie przepływu krwi przez nerki, ostre i choroby przewlekłe nerek, skutki przewlekłej choroby nerek)
B) choroba wątroby (pierwotna i alkoholowa marskość wątroby, zapalenie wątroby, wątrobiak)
C) choroby przewodu pokarmowego i narządów dokrewnych
C) indywidualna nietolerancja niektórych leków (brak enzymów acetylacji – nietolerancja aspiryny)
32. Sposoby i mechanizmy wydalania leków z organizmu. Możliwości zarządzania eliminacją leków.
Sposoby i mechanizmy wydalania leków: eliminacja leków przez wątrobę i nerki oraz niektóre inne narządy:
A) przez nerki przez filtrację, sekrecję, reabsorpcję
B) przez wątrobę przez biotransformację, wydalanie z żółcią
C) przez płuca, ślinę, pot, mleko itp. przez wydzielanie, parowanie
Możliwości zarządzania procesami odstawienia leków:
1. Kontrola pH: w moczu zasadowym zwiększa się wydalanie związków kwaśnych, w moczu kwaśnym wydalanie związków zasadowych
2.stosowanie leków żółciopędnych (cholenzym, allohol)
3. hemodializa, dializa otrzewnowa, hemosorpcja, limfosorpcja
4. Wymuszona diureza (IV NaCl lub glukoza do obciążenia wodą + furosemid lub mannitol)
5. płukanie żołądka, stosowanie lewatyw
33. Pojęcie receptorów w farmakologii, molekularna natura receptorów, mechanizmy sygnalizacyjne działania leków (rodzaje sygnalizacji transbłonowej i mediatory wtórne).
Receptory - Molekularne składniki komórki lub organizmu, które oddziałują z lekami i indukują szereg zdarzeń biochemicznych prowadzących do rozwoju efektu farmakologicznego.
Pojęcie receptorów w farmakologii:
1. Receptory określają ilościowe wzorce działania leku
2. Receptory odpowiadają za selektywność działania leków
3. Receptory pośredniczą w działaniu antagonistów farmakologicznych
Pojęcie receptorów jest podstawą celowego stosowania leków wpływających na procesy regulacyjne, biochemiczne i komunikację.
Molekularna natura receptorów:
1.białka regulatorowe, mediatory działania różnych sygnałów chemicznych: neuroprzekaźniki, hormony, autokoidy
2.enzymy i transbłonowe białka transportujące (Na+, K+ATPaza)
3.białka strukturalne (tubulina, białka cytoszkieletu, powierzchnia komórki)
4.białka jądrowe i kwasy nukleinowe
Mechanizmy sygnalizacyjne działania leku:
1) przenikanie rozpuszczalnych w tłuszczach ligandów przez błonę i ich wpływ na receptory wewnątrzkomórkowe.
2) cząsteczka sygnalizacyjna wiąże się z domeną zewnątrzkomórkową białka transbłonowego i aktywuje aktywność enzymatyczną jego domeny cytoplazmatycznej.
3) cząsteczka sygnalizacyjna wiąże się z kanałem jonowym i reguluje jego otwarcie.
4) cząsteczka sygnalizacyjna wiąże się z receptorem na powierzchni komórki, który jest sprzężony z enzymem efektorowym przez białko G. Białko G aktywuje wtórnego posłańca.
Rodzaje sygnalizacji transbłonowej:
A) przez receptory 1-TMS z aktywnością kinazy tyrozynowej i bez niej
B) przez receptory 7-TMS związane z białkiem G
B) przez kanały jonowe (zależne od liganda, zależne od napięcia, styki szczelinowe)
Pośrednicy wtórni: cAMP, jony Ca2+, DAG, IF3.
34. Fizykochemiczne i chemiczne mechanizmy działania substancji leczniczych.
ALE) Oddziaływanie fizykochemiczne z biosubstratem- działanie nieelektrolityczne.
Główne efekty farmakologiczne: 1) narkotyczne 2) ogólne depresyjne 3) paraliżujące 4) miejscowo drażniące 5) działanie błoniaste.
Charakter chemiczny substancji: chemicznie obojętne węglowodory, etery, alkohole, aldehydy, barbiturany, leki gazowe
Mechanizm działania polega na odwracalnym zniszczeniu błon.
B) Chemiczny(molekularno-biochemiczny) mechanizm działania leków.
Główne rodzaje interakcji chemicznych z biosubstratem:
- Słabe (oddziaływania niekowalencyjne, odwracalne) (wodorowe, jonowe, monodipolowe, hydrofobowe).
- Wiązania kowalencyjne (alkilacja).
Znaczenie niekowalencyjnych interakcji leków: działanie jest niespecyficzne, nie zależy od struktury chemicznej substancji.
Znaczenie kowalencyjnych interakcji leków: akcja jest specyficzna, krytycznie zależy od struktury chemiczne, realizowany jest poprzez oddziaływanie na receptory.
35. Terminy i pojęcia farmakologii ilościowej: efekt, skuteczność, aktywność, agonista (pełny, częściowy), antagonista. Kliniczna różnica między pojęciami działania i skuteczności leków.
 Efekt (odpowiedź)- ilościowa wydajność reakcji interakcji komórki, narządu, układu lub organizmu ze środkiem farmakologicznym.
Efekt (odpowiedź)- ilościowa wydajność reakcji interakcji komórki, narządu, układu lub organizmu ze środkiem farmakologicznym.
Efektywność- miara reakcji wzdłuż osi efektu - wielkość odpowiedzi układu biologicznego na efekt farmakologiczny; Jest to zdolność leków do zapewnienia maksymalnego możliwego efektu.... To znaczy w rzeczywistości jest to maksymalna wielkość efektu, jaki można osiągnąć dzięki wprowadzeniu danego leku. Charakteryzuje się liczbowo wartością Emax. Im wyższy Emax, tym wyższa skuteczność leku.
Działalność- miara wrażliwości na leki wzdłuż osi stężenia, charakteryzuje powinowactwo (powinowactwo liganda do receptora), pokazuje, jaka dawka (stężenie) leku jest zdolna do wywołania efektu standardowego równego 50% maksymalna możliwa dla tego leku. Numerycznie scharakteryzowany wartością EC50 lub ED50. Im wyższa aktywność leku, tym mniejsza jego dawka jest wymagana do odtworzenia efektu terapeutycznego.
Wydajność: 1 = 2> 3
Aktywność: 1> 3> 2
W praktyce klinicznej ważniejsze jest poznanie skuteczności niż aktywności, ponieważ bardziej interesuje nas zdolność leków do wywoływania określonego działania w organizmie.
Agonista- ligand, który wiąże się z receptorem i wywołuje odpowiedź biologiczną, aktywację układu fizjologicznego. Pełen agonista- maksymalna odpowiedź, Częściowy- powodują mniejszą reakcję, nawet gdy wszystkie receptory są zajęte.
Antagonista- ligandy zajmujące receptory lub zmieniające je w taki sposób, że tracą zdolność oddziaływania z innymi ligandami, ale same nie wywołują reakcji biologicznej (blokują działanie agonistów).
 Konkurencyjni antagoniści- oddziałują odwracalnie z receptorami i w ten sposób konkurują z agonistami. Zwiększenie stężenia agonisty może całkowicie wyeliminować działanie antagonisty. Konkurencyjny antagonista przesuwa krzywą dawka-odpowiedź dla agonisty, zwiększa EC50, nie wpływa na Emax.
Konkurencyjni antagoniści- oddziałują odwracalnie z receptorami i w ten sposób konkurują z agonistami. Zwiększenie stężenia agonisty może całkowicie wyeliminować działanie antagonisty. Konkurencyjny antagonista przesuwa krzywą dawka-odpowiedź dla agonisty, zwiększa EC50, nie wpływa na Emax.
Niekonkurencyjni antagoniści- nieodwracalnie zmieniają powinowactwo receptorów do agonisty, często nie dochodzi do wiązania z miejscem aktywnym receptora, wzrost stężenia agonisty nie eliminuje działania antagonisty. Niekompetycyjny antagonista zmniejsza Emax, nie zmienia EC50, a krzywa dawka-odpowiedź jest skompresowana wokół osi pionowej.
36. Ilościowe wzorce działania leków. Prawo zmniejszania odpowiedzi systemów biologicznych. Model Clarka i jego konsekwencje. Ogólny widok koncentracji zależności - efekt we współrzędnych normalnych i lognormalnych.
Model Clarka-Ariensa:
1. Oddziaływanie między ligandem (L) a receptorem (R) jest odwracalne.
2. Wszystkie receptory dla danego ligandu są równoważne i niezależne (ich nasycenie nie wpływa na inne receptory).
3. Efekt jest wprost proporcjonalny do liczby zajętych receptorów.
4. Ligand występuje w dwóch stanach: wolnym i związanym z receptorem.
ALE) ![]() , gdzie Kd jest stałą równowagi, Ke jest aktywnością wewnętrzną.
, gdzie Kd jest stałą równowagi, Ke jest aktywnością wewnętrzną.
B) Ponieważ wraz ze wzrostem liczby ligandów w pewnym momencie wszystkie receptory będą zajęte, wówczas maksymalną możliwą liczbę utworzonych kompleksów ligand-receptor opisuje wzór:
= [R] × ![]() (1)
(1)
Efekt zależy od prawdopodobieństwa aktywacji receptora po związaniu z ligandem, czyli od jego wewnętrznej aktywności (Ke), a więc E = Ke ×. W tym przypadku efekt jest maksymalny przy Ke = 1 i minimalny i Ke = 0. Oczywiście maksymalny efekt opisuje stosunek Emax = Ke ×, gdzie jest całkowitą liczbą receptorów dla danego ligandu
Efekt zależy również od stężenia ligandu na receptorach [C], dlatego
E = Emaks ![]() (2)
(2)
Z powyższych relacji wynika, że EC50 = Kd
![]()
Emax to maksymalny efekt, Bmax to maksymalna liczba związanych receptorów, EC50 to stężenie leku, przy którym występuje efekt równy połowie maksimum, Kd to stała dysocjacji substancji od receptora, przy której 50% receptory są związane.
Prawo malejącej odpowiedzi odpowiada paraboliczna zależność „stężenie - wydajność”. Reakcja na niskie dawki leków zwykle wzrasta wprost proporcjonalnie do dawki... Jednak wraz ze wzrostem dawki wzrost odpowiedzi maleje i ostatecznie można osiągnąć dawkę, przy której nie ma dalszego wzrostu odpowiedzi (ze względu na zajęcie wszystkich receptorów dla danego liganda).
37. Zmiana działania leków. Stopniowa i kwantowa ocena efektu, istoty i zastosowań klinicznych. Środki określić ilościowo aktywność i skuteczność leków w praktyce eksperymentalnej i klinicznej.
Wszystkie efekty farmakologiczne można z grubsza podzielić na dwie kategorie:
ALE) Efekty stopniowe (ciągłe, całkowe)- takie działanie leków, które można zmierzyć ilościowo (działanie leków hipotensyjnych - przez poziom ciśnienia krwi). Opisano stopniową „krzywą dawka-efekt” (patrz str. 36), na podstawie której można oszacować: 1) indywidualną wrażliwość na leki 2) aktywność leku 3) maksymalną skuteczność leku
B) Efekty kwantowe- takie efekty leków, które są wartością dyskretną, znakiem jakościowym, czyli są opisane tylko kilkoma wariantami stanów (ból głowy po zażyciu środka przeciwbólowego, obecny lub nie). Opisano krzywą kwantową dawka-efekt, w której odnotowuje się zależność manifestacji efektu w populacji od wartości przyjętej dawki leku. Wykres dawka-efekt ma kształt kopuły i jest identyczny z krzywą rozkładu normalnego Gaussa. Na podstawie krzywej kwantowej można: 1) ocenić populacyjną wrażliwość leków, 2) odnotować obecność efektu przy danej dawce, 3) wybrać średnią dawkę terapeutyczną.
Różnice między stopniową a kwantową charakterystyką efektu dawki:
Ilościową ocenę aktywności i skuteczności leków przeprowadza się na podstawie konstrukcji krzywych dawka-skutek i ich późniejszej oceny (patrz Rozdział 35)
38. Rodzaje działania leków. Zmiany w działaniu leków po ich ponownym podaniu.
Rodzaje działania leku:
1. Działania lokalne- działanie substancji występującej w miejscu jej podania (znieczulenie - na błonę śluzową)
2. Działania resorpcyjne (systemowe)- działanie substancji, która rozwija się po jej wchłonięciu, wejściu do ogólnego krwiobiegu, a następnie do tkanek. Zależy od dróg podania leków i ich zdolności do przenikania barier biologicznych.
Leki mogą mieć działanie zarówno lokalne, jak i resorpcyjne: Bezpośredni lub Odruch wpływ:
A) bezpośredni wpływ - bezpośredni kontakt z narządem docelowym (adrenalina na sercu).
B) odruch - zmiana funkcji narządów lub ośrodków nerwowych poprzez wpływ na zewnętrzne i interoreceptory (plastry musztardowe z patologią układu oddechowego odruchowo poprawiają ich trofizm)
Zmiany w działaniu leków po ich ponownym wprowadzeniu:
1. Kumulacja- wzrost efektu z powodu nagromadzenia leków w organizmie:
a) kumulacja materiałowa – akumulacja substancji czynnej w organizmie (glikozydy nasercowe)
b) kumulacja funkcjonalna – nasilające się zmiany funkcji układów organizmu (zmiany funkcji ośrodkowego układu nerwowego w przewlekłym alkoholizmie).
2. Tolerancja (uzależnienie) - Zmniejszenie reakcji organizmu na powtarzające się zastrzyki leków; w celu przywrócenia reakcji na leki należy go podawać w coraz większych dawkach (diazepam):
A) prawdziwa tolerancja - obserwowana zarówno przy dojelitowym, jak i pozajelitowym podawaniu leków, nie zależy od stopnia jej wchłaniania do krwiobiegu. Opiera się na farmakodynamicznych mechanizmach uzależnienia:
1) odczulanie - zmniejszenie wrażliwości receptora na lek (agoniści receptorów b-adrenergicznych, przy długotrwałym stosowaniu prowadzą do fosforylacji receptorów b-adrenergicznych, które nie są w stanie odpowiedzieć na agonistów receptorów b-adrenergicznych)
2) Regulacja w dół - zmniejszenie liczby receptorów leku (przy wielokrotnym podawaniu narkotycznych leków przeciwbólowych liczba receptorów opioidowych maleje i do wywołania pożądanej odpowiedzi wymagane są coraz większe dawki leku). Jeśli lek blokuje receptory, mechanizm tolerancji na niego może być związany z regulacją w górę - wzrostem liczby receptorów leku (b-blokerów)
3) włączenie kompensacyjnych mechanizmów regulacji (przy wielokrotnych wstrzyknięciach leków przeciwnadciśnieniowych zapaść występuje znacznie rzadziej niż przy pierwszym podaniu z powodu adaptacji baroreceptorów)
B) tolerancja względna (pseudotolerancja) - rozwija się tylko wraz z wprowadzeniem leków do środka i wiąże się ze spadkiem szybkości i kompletności wchłaniania leku
3. Tachyfilaksja- stan, w którym częste podawanie leków powoduje rozwój tolerancji już po kilku godzinach, ale przy dość rzadkim podawaniu leków jego działanie jest w pełni zachowane. Rozwój tolerancji jest zwykle związany z wyczerpywaniem się układów efektorowych.
4. Uzależnienie od narkotyków- nieodparta chęć zażycia wcześniej podanej substancji. Przydziel psychiczne (kokaina) i fizyczne (morfina) uzależnienie od narkotyków.
5. Nadwrażliwość- reakcja alergiczna lub inna reakcja immunologiczna na leki podawane wielokrotnie.
39. Zależność działania leków od wieku, płci i Cechy indywidulane organizm. Znaczenie rytmów dobowych.
ALE) Od wieku: u dzieci i osób starszych zwiększona wrażliwość na leki (ponieważ dzieci mają niedobór wielu enzymów, czynność nerek, zwiększona przepuszczalność BBB, w starszym wieku wchłanianie leków jest spowolnione, metabolizm jest mniej wydajny, szybkość wydalania leki przez nerki są zmniejszone):
|
1. Noworodki mają zmniejszoną wrażliwość na glikozydy nasercowe, ponieważ mają więcej Na + / K + -ATPaz (cele działania glikozydów) na jednostkę powierzchni kardiomiocytu. 2. Dzieci mają niższą wrażliwość na sukcynylocholinę i atrakurię, ale zwiększoną na wszystkie inne środki zwiotczające mięśnie. 3. Leki psychotropowe mogą powodować nieprawidłowe reakcje u dzieci: psychostymulanty – mogą zwiększać koncentrację i zmniejszać nadpobudliwość ruchową, uspokajające – wręcz przeciwnie, mogą powodować tzw. nietypowe pobudzenie. |
1. Wrażliwość na glikozydy nasercowe gwałtownie wzrasta z powodu zmniejszenia liczby ATPaz Na + / K +. 2. Zmniejsza wrażliwość na b-blokery. 3. Wrażliwość na blokery kanału wapniowego wzrasta, ponieważ baroreflex jest osłabiony. 4. Istnieje nietypowa reakcja na leki psychotropowe, podobna do reakcji dzieci. |
B) Z podłogi:
1) leki przeciwnadciśnieniowe - klonidyna, beta-blokery, leki moczopędne mogą powodować naruszenie funkcje seksualne u mężczyzn, ale nie wpływają na pracę układ rozrodczy kobiety.
2) sterydy anaboliczne są bardziej skuteczne u kobiet niż u mężczyzn.
W) Z indywidualnych cech organizmu: niedobór lub nadmiar niektórych enzymów metabolizmu leków prowadzi do nasilenia lub osłabienia ich działania (niedobór pseudocholinoesterazy we krwi - nieprawidłowo przedłużone rozluźnienie mięśni przy stosowaniu sukcynylocholiny)
G) Z rytmów dobowych: zmiana wpływu leków na organizm, ilościowo i jakościowo, w zależności od pory dnia (maksymalny efekt przy maksymalnej aktywności).
40. Zmienność i zmienność działania leków. Hipo - i nadreaktywność, tolerancja i tachyfilaksja, nadwrażliwość i idiosynkrazja. Przyczyny zmienności działania leków i racjonalnej strategii terapii.
Zmienność odzwierciedla różnice między osobami w odpowiedzi na dany lek.
Przyczyny zmienności działania leku:
1) zmiana stężenia substancji w strefie receptorowej - ze względu na różnice w szybkości wchłaniania, jej dystrybucji, metabolizmie, eliminacji
2) wahania stężenia endogennego ligandu receptora – propranololu (β-blokera) spowalniają tętno u osób z podwyższonym poziomem katecholamin we krwi, ale nie wpływają na tętno podstawowe u sportowców.
3) zmiany gęstości lub funkcji receptorów.
4) zmiany w składnikach reakcji zlokalizowanych dystalnie do receptora.
Racjonalna strategia terapii: mianowanie i dawkowanie leków, biorąc pod uwagę powyższe przyczyny zmienności działania leku.
Hiporeaktywność- zmniejszenie działania danej dawki leków w porównaniu z efektem obserwowanym u większości pacjentów. Nadreaktywność- wzrost działania danej dawki leków w porównaniu z efektem obserwowanym u większości pacjentów.
Tolerancja, tachyfilaksja, nadwrażliwość – patrz punkt 38
Idiosynkrazja- wypaczona reakcja organizmu na dany lek, związana z genetyczną charakterystyką metabolizmu leku lub z indywidualną reaktywnością immunologiczną, w tym z reakcjami alergicznymi.
41. Ocena bezpieczeństwa leków. Indeks terapeutyczny i standardowe marginesy bezpieczeństwa.
Ocena bezpieczeństwa przeprowadzana jest na dwóch poziomach:
A) przedkliniczne (uzyskanie informacji o toksyczności leków, wpływie na funkcje rozrodcze embriotoksyczność i teratogenność, skutki długoterminowe)
B) kliniczne (dalsza ocena skuteczności i bezpieczeństwa leków)
Jeśli po osiągnięciu plateau efektu dawka leku będzie nadal wzrastać, po pewnym czasie zacznie się objawiać jego toksyczny efekt. Zależność działania toksycznego od dawki (stężenia) leku ma ten sam charakter, co jego korzystne działanie i można ją opisać krzywymi stopniowymi lub kwantowymi. Te krzywe można również wykorzystać do określenia wartości TD50 lub TC50- dawka toksyczna (stężenie) leków, która powoduje efekt toksyczny równy 50% maksimum (dla krzywej kwantowej - efekt toksyczny u 50% osobników w populacji). Czasami zamiast TD50 używają wskaźnika LD50 - dawka śmiertelna, co powoduje śmierć 50% obiektów w populacji.
Ocena bezpieczeństwa leku jest charakteryzowana na podstawie krzywych gradientowych lub kwantowych krzywych dawka-efekt oraz następujących wskaźników:
ALE) Indeks terapeutyczny Czy stosunek dawek toksycznych i skutecznych leku, które powodują pojawienie się efektu połowy maksymalnego: TI = TD50 / ED50. Im wyższa wartość indeksu terapeutycznego, tym lek jest bezpieczniejszy.
B) Szerokość terapeutyczna (okno terapeutyczne) Jest to zakres dawek między minimalną dawką terapeutyczną a minimalną dawką toksycznych leków. Jest to bardziej poprawny wskaźnik bezpieczeństwa leku, ponieważ pozwala na uwzględnienie stopnia nasilenia działań niepożądanych na krzywej dawka-skutek.
W) Niezawodny współczynnik bezpieczeństwa- Jest to stosunek minimalnej dawki toksycznej do maksymalnej dawki skutecznej (PNF = TD1 / ED99), pokazuje, ile razy można przekroczyć dawkę terapeutyczną leku bez ryzyka zatrucia (niepożądane skutki).
G) Korytarz terapeutyczny Jest to zakres skutecznych stężeń leku we krwi, które muszą zostać wytworzone i utrzymane w organizmie, aby osiągnąć pożądany efekt terapeutyczny.
42,46. Interakcja leków. Niekompatybilność leków (ponieważ pytania są ze sobą powiązane, wybierz w zależności od okoliczności)
Interakcje pomiędzy lekami- jest to zmiana nasilenia i charakteru skutków przy jednoczesnym lub wstępnym stosowaniu kilku leków.
Powody niechcianych interakcji:
1) polipragmazja – 6 lub więcej leków daje 7 razy więcej skutków ubocznych niż w przypadku, gdy leków jest mniej niż 6.
2) błędy lekarzy
3) naruszenie schematu dawkowania
Uzasadnienie terapii skojarzonej:
1. Monoterapia nie jest wystarczająco skuteczna.
2. Brak terapii etiotropowej w większości chorób Þ potrzeba oddziaływania leczniczego na różne ogniwa patogenezy
3. Wielochorobowość - niż starszy człowiek, tym więcej ma chorób, które występują w tym samym czasie
4. Konieczność korygowania niepożądanych skutków leków
5. Zmniejszenie liczby przyjęć i podawania leków (wygoda dla pacjenta, oszczędność pracy pracowników służby zdrowia)
Rodzaje interakcji:
i. Interakcja farmaceutyczna - Rodzaj interakcji związany z reakcją fizykochemiczną między lekami podczas wytwarzania produktu leczniczego, jeszcze przed wprowadzeniem tych środków do organizmu człowieka
A) typowe błędy prowadzące do niezgodności farmaceutycznej: nie uwzględnia się wypisywania skomplikowanych recept, niewłaściwego przechowywania, możliwości adsorpcji leków na powierzchni plastiku (azotanów organicznych)
B) problemy z terapią infuzyjną: mieszanie rozpuszczalnych soli, pochodnych nierozpuszczalnych słabych kwasów lub zasad prowadzi do ich wytrącania; w płynnych postaciach dawkowania glikozydy nasercowe i alkaloidy ulegają hydrolizie, AB ulega zniszczeniu; pH podłoża (alkaloidy wytrącają się w środowisku alkalicznym)
C) zalecenia: 1) Wszystkie mieszaniny lepiej przygotować ex tempore 2) Najbardziej niezawodnym rozwiązaniem jest jeden lek 3) Przed użyciem należy sprawdzić wszystkie roztwory pod kątem zawiesiny 4) Interakcja może zachodzić bez widocznych zmian w roztworach 5) Leki nie mogą dodać do krwi i roztworów AK 6) W przypadku braku specjalnych instrukcji preparaty należy rozpuścić w 5% roztworze glukozy (pH 3,5-6,5), izotonicznym roztworze NaCl (pH 4,5-7,0).
Roztwór glukozy stabilizowany HCl jest niezgodny z epinefryną, benzylopenicyliną, apomorfiną, kanamycyną, witaminą C, oleandomycyną, glikozydami nasercowymi. Glikozydy nasercowe są niezgodne z atropiną, papaweryną, platifillin. AB są niezgodne z heparyną, hydrokortyzonem. Witaminy z grupy B są ze sobą niekompatybilne, z witaminami PP, C. Witaminy PP i C również są ze sobą niekompatybilne.
Nie można mieszać z innymi lekami: fenotiazydem, chlorpromazyną, barbituranami, preparatami witaminy C, amfoterycyną B, furosemidem, sulfadiazyną, aminofiliną, adrenomimetykami.
II... Farmakologiczny- interakcja leków, która objawia się dopiero w ludzkim ciele po ich łącznym zastosowaniu
A) farmakokinetyka
1) podczas fazy ssania.
Przy wprowadzaniuZa Osinterakcja jest określona przez:
1. kwasowość środowiska
2.bezpośrednie oddziaływanie w przewodzie pokarmowym
Tetracykliny oddziałują z wapniem, glinem, żelazem, magnezem, tworząc kompleksy chelatowe. Cholestyramina zaburza wchłanianie pochodnych kwasowych, preparatów wapniowych, warwaryny, digoksyny, digitoksyny, witamin rozpuszczalnych w tłuszczach, trimetoprimu, klindamycyny, cefaleksyny, tetracykliny. Preparaty żelaza są lepiej wchłaniane z witaminą C. Preparaty żelaza z węglanami, tetracyklinami są słabo wchłaniane.
3.Ruchliwość przewodu pokarmowego
Spowalniają perystaltykę: niektóre leki przeciwdepresyjne, leki przeciwhistaminowe, leki przeciwpsychotyczne fenotiazynowe, środki odurzające, zwiększają wchłanianie digoksyny, kortykosteroidy, leki przeciwzakrzepowe, zmniejszają wchłanianie lewodopy. Wzmacniają perystaltykę i zwiększają ewakuację z przewodu pokarmowego: metoklopramid, środki przeczyszczające. Zmniejszenie wchłaniania leków: fenobarbital – gryzeofulwina, aspiryna – indometacyna i diklofenak, PASK – ryfampicyna.
Metody kontrolowania wchłaniania podczas podawania pozajelitowego: znieczulenie miejscowe + epinefryna + fenylefryna - zmniejsza się wchłanianie środków miejscowo znieczulających
4.flora jelitowa
5. zmiana mechanizmu ssącego
2) przy dystrybucji i deponowaniu:
1.bezpośrednie oddziaływanie w osoczu krwi: gentamycyna + ampicylina lub karbenicylina - zmniejszają aktywność gentamycyny
2. konkurencyjne wypieranie z połączenia z albuminą w osoczu krwi: indometacyna, digitoksyna, warfaryna są związane z białkami krwi o 90-98%, dlatego dwukrotny wzrost wolnej frakcji leków jest gwałtownym wzrostem działania toksycznego; NLPZ zastępują: warfarynę, fenytoinę, metotreksat.
Determinanty decydujące o znaczeniu klinicznym tej interakcji:
ü Wartość Vd (duża - bez problemu, mała - możliwa)
ü wpływ jednej substancji leczniczej na aktywność mechanizmów transportowych poprzez mechanizmy innych leków: transport leku wzrasta w zależności od dawki - insuliny, ACTH, angiotensyny, kinin itp .; insulina zwiększa stężenie izoniazydu tylko w płucach, a bawełnianoromazyny tylko w SMC.
3. Przemieszczenie z wiązania białek tkankowych: chinidyna wypiera digoksynę + zmniejsza wydalanie nerkowe, zwiększając tym samym ryzyko toksyczności digoksyny
3) w procesie metabolizmu
Leki mogą zwiększać lub zmniejszać aktywność cytochromu P450 i jego enzymów (etanol zwiększa aktywność niektórych izoenzymów cytochromu)
Często oddziałujące inhibitory enzymów:
1. AB: cyprofloksacyna, erytromycyna, izoniazyd, metronidazol
2. Leki sercowo-naczyniowe: amiodaron, diltiazem, chinidyna, werapamil
3. Leki przeciwdepresyjne: fluoksetyna, sertralen
4. Leki przeciwwydzielnicze: cymetydyna, omeprazol
5. Leki przeciwreumatyczne: allopurynol
6. Fungicydy: flukonazol, intrakanazol, ketokonazol, mikonazol
7. Leki przeciwwirusowe: indynawir, retonawir, sakwinawir
8. Inne: disulfiram, walproinian sodu
Leki dające efekty toksyczne w hamowaniu MAO: adrenomimetyki, sympatykomimetyki, leki przeciwparkinsonowskie, narkotyczne leki przeciwbólowe, fenotiazyny, środki uspokajające, przeciwnadciśnieniowe leki moczopędne, leki hipoglikemizujące
4) W trakcie wylęgu- ponad 90% leków jest wydalane z moczem.
Wpływ na pH moczu i stopień jonizacji leków, ich lipofilność i reabsorpcję
1. interakcja podczas dyfuzji biernej: część leku jest wydalana w postaci niezmienionej, część leku jest jonizowana przy pH moczu 4,6-8,2. Alkalizacja moczu ma znaczenie kliniczne: zatrucie kwasem acetylosalicylowym lub fenobarbitalem, podczas przyjmowania sulfonamidów (zmniejszenie ryzyka krystalurii), przyjmowanie chinidyny. Zwiększona kwasowość moczu: zwiększone wydalanie amfetaminy (praktyczne znaczenie dla wykrywania tego leku u sportowców)
2.interakcja w okresie transportu aktywnego: probenezyd + penicylina wydłuża czas trwania ruchu penicyliny, probenecyd + salicylany - eliminacja działania urykozurycznego probenecydu, penicylina + CA - zmniejszenie wydalania penicyliny
Wpływ składu moczu na wydalanie leku:
Wzrost cukru w moczu - wzrost wydalania: witaminy C, chloramfenikolu, morfiny, izoniazydu, glutationu i ich metabolitów.
B) farmakodynamika Czy interakcja leków wiąże się ze zmianą farmakodynamiki jednego z nich pod wpływem drugiego (pod wpływem hormonów tarczycy wzrasta synteza receptorów b-adrenergicznych w mięśniu sercowym i zwiększa się wpływ adrenaliny na mięsień sercowy ).
Przykłady klinicznie istotnych niepożądanych interakcji synergicznych:
NLPZ + varvarina – zwiększone ryzyko krwawienia
Alkohol + benzodiazepiny - nasilenie działania uspokajającego
Inhibitory ACE + K + - oszczędzające leki moczopędne - zwiększone ryzyko hiperkaliemii
Werapamil + b-blokery - bradykardia i asystolia
Alkohol jest silnym induktorem enzymów mikrosomalnych, prowadzi do rozwoju tolerancji na leki (zwłaszcza na środki znieczulające i nasenne), zwiększa ryzyko uzależnienia od leków.
43. Interakcja leków. Antagonizm, synergia, ich rodzaje. Charakter zmiany działania leków (aktywność, skuteczność) w zależności od rodzaju antagonizmu.
W wyniku interakcji leków mogą wystąpić następujące stany: a) nasilenie działania kombinacji leków b) osłabienie działania kombinacji leków c) niezgodność leków
Wzmocnienie efektów kombinacji leków realizowane jest na trzy sposoby:
1) Podsumowanie efektów lub interakcji addytywnej- pogląd interakcje leków w którym efekt połączenia jest równy prostej sumie efektów każdego z leków oddzielnie. Tj. 1+1=2 ... Jest to charakterystyczne dla leków z jednej grupy farmakologicznej, które mają wspólny cel działania (działanie neutralizujące kwasy kombinacji wodorotlenku glinu i magnezu jest równe sumie ich zdolności neutralizacji kwasów osobno)
2) synergizm – rodzaj interakcji, w której efekt połączenia przekracza sumę efektów każdej z substancji wziętych oddzielnie. Tj. 1+1=3 ... Synergia może dotyczyć zarówno pożądanego (terapeutycznego), jak i niepożądanego działania leków. Łączne podawanie dichlorotiazydu tiazydowego moczopędnego i inhibitora ACE enalaprilu prowadzi do nasilenia działania hipotensyjnego każdego z leków stosowanych w leczeniu nadciśnienia tętniczego. Jednak jednoczesne podawanie antybiotyków aminoglikozydowych (gentamycyny) i diuretycznego furosemidu pętlowego powoduje gwałtowny wzrost ryzyka działania ototoksycznego i rozwoju głuchoty.
3) wzmocnienie to rodzaj interakcji lekowej, w której jeden z leków, który sam w sobie nie ma takiego działania, może prowadzić do gwałtownego nasilenia działania innego leku. Tj. 1+0=3 (kwas klawulanowy nie ma działania przeciwdrobnoustrojowego, ale może wzmocnić działanie antybiotyku b-laktamowego, amoksycyliny, ponieważ blokuje b-laktamazę; adrenalina nie działa miejscowo znieczulająco, ale po dodaniu do roztwór ultrakainy, znacznie wydłuża działanie znieczulające poprzez spowolnienie wchłaniania środka znieczulającego z miejsca wstrzyknięcia).
Efekty tłumiące Leki stosowane razem nazywane są antagonizmem:
1) Antagonizm chemiczny lub antidotum- chemiczne oddziaływanie substancji ze sobą z tworzeniem nieaktywnych produktów (chemiczny antagonista jonów żelaza deferoksamina, który wiąże je w nieaktywne kompleksy; siarczan protaminy, którego cząsteczka ma nadmiar dodatniego ładunku - chemiczny antagonista heparyny, cząsteczka z czego ma nadmiar ładunku ujemnego). U podstaw działania odtrutek (odtrutek) leży antagonizm chemiczny.
2) Antagonizm farmakologiczny (bezpośredni)- antagonizm wywołany wielokierunkowym działaniem 2 substancji leczniczych na te same receptory w tkankach. Antagonizm farmakologiczny może być konkurencyjny (odwracalny) i niekonkurencyjny (nieodwracalny):
A) antagonizm konkurencyjny: konkurencyjny antagonista odwracalnie wiąże się z aktywnym centrum receptora, tj. osłania go przed działaniem agonisty. Ponieważ stopień wiązania substancji z receptorem jest proporcjonalny do stężenia tej substancji, efekt konkurencyjnego antagonisty można przezwyciężyć przez zwiększenie stężenia agonisty. Wyprze antagonistę z miejsca aktywnego receptora i wywoła pełną odpowiedź tkankową. To. antagonista kompetycyjny nie zmienia maksymalnego działania agonisty, ale dla interakcji agonisty z receptorem wymagane jest wyższe stężenie. Antagonista konkurencyjny Przesuwa krzywą dawka-odpowiedź dla agonisty w prawo w stosunku do wartości początkowych i zwiększa EC50 dla agonisty bez wpływu na wartość E Maks..
W praktyce medycznej często stosuje się antagonizm konkurencyjny. Ponieważ efekt konkurencyjnego antagonisty można przezwyciężyć, jeśli jego stężenie spadnie poniżej poziomu agonisty, podczas leczenia konkurencyjnymi antagonistami konieczne jest stałe utrzymywanie jego poziomu na wystarczająco wysokim poziomie. Innymi słowy, efekt kliniczny konkurencyjnego antagonisty będzie zależał od jego okresu półtrwania w fazie eliminacji i stężenia pełnego agonisty.
B) antagonizm niekonkurencyjny: antagonista niekonkurencyjny wiąże się prawie nieodwracalnie z aktywnym centrum receptora lub ogólnie oddziałuje z jego centrum allosterycznym. Dlatego bez względu na to, jak wzrasta stężenie agonisty, nie jest on w stanie wyprzeć antagonisty z jego połączenia z receptorem. Ponieważ niektóre receptory związane z niekonkurencyjnym antagonistą nie są już zdolne do aktywacji , wartość EMaks. zmniejsza się, powinowactwo receptora do agonisty nie zmienia się, więc wartość EC50 pozostaje taka sama. Na krzywej dawka-skutek działanie niekonkurencyjnego antagonisty objawia się kompresją krzywej względem osi pionowej bez przesuwania jej w prawo.
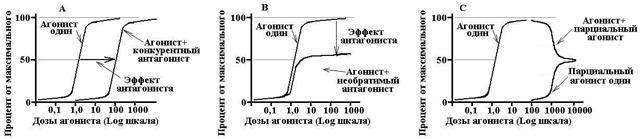
Schemat 9. Rodzaje antagonizmu.
A - konkurencyjny antagonista przesuwa krzywą dawka-efekt w prawo, to znaczy zmniejsza wrażliwość tkanki na agonistę bez zmiany jego działania. B - niekonkurencyjny antagonista zmniejsza wielkość odpowiedzi tkankowej (efektu), ale nie wpływa na jej wrażliwość na agonistę. C - wariant zastosowania częściowego agonisty na tle pełnego agonisty. Wraz ze wzrostem stężenia częściowy agonista wypiera pełnego z receptorów iw rezultacie odpowiedź tkankowa spada od maksymalnej odpowiedzi na pełnego agonistę do maksymalnej odpowiedzi na częściowego agonistę.
W praktyce medycznej rzadziej stosuje się antagonistów niekonkurencyjnych. Z jednej strony mają niewątpliwą zaletę, ponieważ ich działanie nie może zostać przezwyciężone po związaniu z receptorem, a zatem nie zależy od okresu połowicznej eliminacji antagonisty ani od poziomu agonisty w organizmie. Efekt niekonkurencyjnego antagonisty będzie determinowany jedynie szybkością syntezy nowych receptorów. Ale z drugiej strony, jeśli dojdzie do przedawkowania tego leku, niezwykle trudno będzie wyeliminować jego działanie.
|
Konkurencyjny antagonista |
Niekonkurencyjny antagonista |
|
Podobny w strukturze do agonisty |
Strukturalnie różni się od agonisty |
|
Wiąże się z aktywnym centrum receptora |
Wiąże się z miejscem allosterycznym receptora |
|
Przesuwa krzywą dawka-odpowiedź w prawo |
Przesuwa w pionie krzywą dawka-odpowiedź |
|
Antagonista zmniejsza wrażliwość tkanek na agonistę (EC50), ale nie wpływa na maksymalny efekt (Emax), który można osiągnąć przy wyższym stężeniu. |
Antagonista nie zmienia wrażliwości tkanek na agonistę (EC50), ale zmniejsza wewnętrzną aktywność agonisty i maksymalną odpowiedź tkankową na niego (Emax). |
|
Działanie antagonistyczne można odwrócić dużą dawką agonisty |
Efektu antagonistycznego nie można odwrócić przy dużej dawce agonisty. |
|
Działanie antagonisty zależy od stosunku dawki agonisty do antagonisty |
Działanie antagonisty zależy tylko od jego dawki. |
Losartan jest kompetycyjnym antagonistą receptorów AT1 angiotensyny, zakłóca interakcję angiotensyny II z receptorami i pomaga obniżyć ciśnienie krwi. Skutki losartanu można przezwyciężyć, podając dużą dawkę angiotensyny II. Walsartan jest niekonkurencyjnym antagonistą tych samych receptorów AT1. Jej działania nie da się przezwyciężyć nawet przy podawaniu dużych dawek angiotensyny II.
Interesująca jest interakcja zachodząca między pełnymi i częściowymi agonistami receptora. Jeżeli stężenie pełnego agonisty przekracza poziom częściowego, to w tkance obserwuje się maksymalną odpowiedź. Jeśli poziom częściowego agonisty zaczyna rosnąć, wypiera pełnego agonistę z wiązania z receptorem, a odpowiedź tkankowa zaczyna spadać od maksimum dla pełnego agonisty do maksimum dla częściowego agonisty (tj. poziomu, przy którym zajmuje wszystkie receptory).
3) Fizjologiczny (pośredni) antagonizm- antagonizm związany z wpływem 2 substancji leczniczych na różne receptory (cele) w tkankach, co prowadzi do wzajemnego osłabienia ich działania. Na przykład obserwuje się antagonizm fizjologiczny między insuliną a adrenaliną. Insulina aktywuje receptory insuliny, dzięki czemu transport glukozy do komórki wzrasta, a poziom glikemii spada. Epinefryna aktywuje receptory b2-adrenergiczne wątroby, mięśni szkieletowych oraz stymuluje rozpad glikogenu, co ostatecznie prowadzi do wzrostu poziomu glukozy. Ten rodzaj antagonizmu jest często stosowany w nagłych wypadkach u pacjentów z przedawkowaniem insuliny, które doprowadziło do śpiączki hipoglikemicznej.
44. Skutki uboczne i toksyczne leków. Teratogenne, embriotoksyczne, mutagenne działanie leków.
Skutki uboczne- te efekty, które występują, gdy substancje są stosowane w dawkach terapeutycznych i stanowią spektrum ich działania farmakologicznego (przeciwbólowa morfina w dawkach terapeutycznych powoduje euforię) mogą być pierwotne i wtórne:
A) pierwotne skutki uboczne - jako bezpośrednia konsekwencja działania tego leku na określony substrat (hiposaliwacja podczas stosowania atropiny w celu wyeliminowania bradyarytmii)
B) wtórne skutki uboczne – pośrednio występujące działania niepożądane (AB, tłumienie prawidłowej mikroflory, może prowadzić do nadkażenia)
Efekty toksyczne- działania niepożądane objawiające się tym lekiem, gdy opuszcza on zakres terapeutyczny (przedawkowanie leku)
Selektywność działania leku zależy od jego dawki. Im wyższa dawka leku, tym mniej selektywny staje się.
Działanie teratogenne- zdolność leków podawanych kobiecie w ciąży do wywoływania anatomicznych nieprawidłowości rozwoju płodu (talidomid: fokomelia, leki przeciwblastoma: liczne wady)
Działanie embriotoksyczne- działanie niepożądane niezwiązane z naruszeniem organogenezy w pierwszych trzech miesiącach ciąży. Wydaje się, że w późniejszym terminie Fetotoksyczne działanie.
Działanie mutagenne leków- uszkodzenie komórki rozrodczej i jej aparatu genetycznego leków, objawiające się zmianą genotypu potomstwa (adrenalina, cytostatyki).
Działanie rakotwórcze leków- zdolność niektórych leków do indukowania kancerogenezy.
45. Medyczne i społeczne aspekty zwalczania narkomanii, narkomanii i alkoholizmu. Pojęcie nadużywania substancji.
« Jest mało prawdopodobne, że ludzkość jako całość kiedykolwiek obejdzie się bez sztucznego raju. Większość mężczyzn i kobiet prowadzi tak bolesne życie, że najlepszy przypadek tak monotonna, nędzna i ograniczona, że chęć „oddalenia się” od niej, rozłączenia się chociaż na kilka chwil, jest i zawsze była jedną z głównychŻyczę Nie ma duszy„(Huxley, praca „Drzwi percepcji”)
1) Uzależnienie od narkotyków- stan umysłu i/lub stan fizyczny, który jest konsekwencją działania leków na organizm i charakteryzuje się specyficznymi reakcjami behawioralnymi, trudno jest przezwyciężyć chęć ponownego zażywania leków w celu osiągnięcia szczególnego stanu psychicznego efekt lub w celu uniknięcia dyskomfortu w przypadku braku leków w ciele. Uzależnienie od narkotyków charakteryzuje się:
ALE) Uzależnienie psychologiczne- rozwój niepokoju emocjonalnego po zaprzestaniu zażywania narkotyków. Człowiek czuje się pusty, popada w depresję, odczuwa strach, niepokój, jego zachowanie staje się agresywne. Wszystkie te objawy psychopatologiczne powstają na tle myśli o potrzebie wstrzykiwania sobie narkotyków, które spowodowały uzależnienie. Chęć zażywania narkotyków może wahać się od zwykłego pragnienia do namiętnego pragnienia zażywania narkotyków, które pochłania wszystkie inne potrzeby i zamienia się w sens życia danej osoby. Uważa się, że uzależnienie psychiczne rozwija się, gdy dana osoba uświadamia sobie, że może osiągnąć optymalne samopoczucie wyłącznie poprzez wprowadzenie leków. Podstawą uzależnienia psychicznego jest wiara osoby w działanie leku (w literaturze opisano przypadki rozwoju uzależnienia psychicznego od placebo).
B) Uzależnienie fizyczne- naruszenie normalnego stanu fizjologicznego organizmu, które wymaga stałej obecności w nim leków w celu utrzymania stanu równowagi fizjologicznej. Odstawienie leku powoduje rozwój specyficznego zespołu objawów - zespołu abstynencyjnego - zespołu zaburzeń psychicznych i neurowegetatywnych w postaci dysfunkcji w kierunku przeciwnym do charakterystycznego dla działania (morfina likwiduje ból, hamuje ośrodek oddechowy, zwęża źrenice, powoduje zaparcia, przy objawach odstawiennych u pacjenta pojawia się rozdzierający ból, częsty głośny oddech, rozszerzenie źrenic i uporczywa biegunka)
W) Tolerancja... Tolerancja na leki powodujące uzależnienie od narkotyków jest często przekrojowa, to znaczy powstaje nie tylko na dany związek chemiczny, ale także na wszystkie związki strukturalnie podobne. Na przykład u pacjentów uzależnionych od morfiny pojawia się tolerancja nie tylko na nią, ale także na inne opioidowe leki przeciwbólowe.
Dla rozwoju uzależnienia od narkotyków obecność wszystkich 3 kryteriów nie jest warunkiem koniecznym; Tabela 3 przedstawia główne rodzaje uzależnienia od narkotyków i jego elementy składowe.
Opioidy, barbiturany, alkohol powodują silne uzależnienie fizyczne i psychiczne oraz tolerancję. Leki przeciwlękowe (diazepam, alprazolam) powodują głównie uzależnienie psychiczne.
2) Uzależnienie (uzależnienie od narkotyków)- Jest to niezwykle ciężka forma uzależnienia, kompulsywne zażywanie narkotyków, charakteryzujące się stale narastającą, nieodpartą chęcią podawania tego leku, zwiększania jego dawki. Kompulsywność pożądania oznacza, że potrzeba podania leku przez pacjenta dominuje nad wszystkimi innymi (nawet żywotnymi) potrzebami. Z punktu widzenia tej definicji głód morfiny to uzależnienie od narkotyków, podczas gdy głód nikotynowy to uzależnienie od narkotyków.
3) Uzależniony od medycyny- charakteryzuje mniej intensywne pragnienie leków, gdy odmowa przyjmowania leków powoduje jedynie uczucie łagodnego dyskomfortu, bez rozwoju uzależnienia fizycznego lub szczegółowego obrazu uzależnienia psychicznego. To. uzależnienie obejmuje tę część uzależnienia od narkotyków, która nie pasuje do definicji uzależnienia. Na przykład wspomniane uzależnienie od nikotyny jest formą uzależnienia.
4) Narkomania- nieautoryzowane zażywanie leków w takich dawkach i w sposób, który odbiega od przyjętych standardów medycznych lub społecznych w danej kulturze i czasie. To. nadużywanie narkotyków obejmuje jedynie społeczne aspekty używania narkotyków. Przykładem nadużycia jest stosowanie sterydów anabolicznych w sporcie lub w celu poprawy sylwetki młodych mężczyzn.
5) Alkoholizm- przewlekłe nadużywanie alkoholu (alkoholu etylowego), prowadzące obecnie do uszkodzenia wielu narządów (wątroby, przewodu pokarmowego, ośrodkowego układu nerwowego, układu krążenia, odpornościowego) i połączone z uzależnieniem psychofizycznym.
6) Nadużywanie substancji- przewlekłe nadużywanie różnych narkotyków (m.in. narkotyki, alkohol, halucynogeny), objawiające się różnymi zaburzeniami psychicznymi i somatycznymi, zaburzeniami zachowania, degradacją społeczną.
Leczenie uzależnienia od narkotyków trudne i niewdzięczne zadanie. Do tej pory nie stworzono skutecznej metody, która zapewniłaby powodzenie leczenia u ponad 30-40% pacjentów. Osiągnięcie jakichkolwiek zauważalnych rezultatów jest możliwe tylko przy pełnej współpracy wysiłków pacjenta, lekarza i środowiska społecznego, w którym znajduje się chory (zasada dobrowolności i indywidualności). Nowoczesne techniki opierają się na następujących zasadach:
ü metody psychoterapeutyczne i terapii zajęciowej;
ü grupowe leczenie i rehabilitacja (towarzystwo anonimowych alkoholików, narkomanów)
ü stopniowe lub nagłe odstawienie leku na tle terapii detoksykacyjnej
ü prowadzenie terapii substytucyjnej (zastąpienie środka odurzającego analogami o powolnym i długotrwałym działaniu z późniejszym ich anulowaniem; np. program tzw. metadonowej terapii substytucyjnej dla osób uzależnionych od heroiny)
ü leczenie specyficznymi antagonistami (nalokson i naltrekson) lub środkami uczulającymi (teturam)
ü neurochirurgiczne metody kriodestrukcji zakrętu obręczy i hipokampa
47. Rodzaje farmakoterapii. Deontologiczne problemy farmakoterapii.
Farmakoterapia (FT) - zestaw metod leczenia opartych na stosowaniu leków. Główne typy FT:
1.etiotropowe PT – korekta i eliminacja przyczyny choroby (AB w chorobach zakaźnych)
2.patogenetyczne FT – wpływ na mechanizm rozwoju choroby (inhibitory ACE w nadciśnieniu)
3.objawowe FT – eliminacja objawów choroby, gdy nie można wpłynąć na jej przyczynę lub patogenezę (NLPZ na grypę)
4.Substytucja FT – stosowanie leków w przypadku niedoboru naturalnych substancji biologicznie czynnych (insulina w cukrzycy)
5.profilaktyczny FT (szczepionki, surowice, kwas acetylosalicylowy na chorobę niedokrwienną serca)
Stosunek społeczeństwa do narkotyków dalej obecny etap : 1) chęć uzyskania korzyści bez ryzyka 2) nadzieja na cud, wizje 3) brak zrozumienia ryzyka zażywania narkotyków 4) oburzenie i „sprawiedliwe oburzenie”, pospieszne oceny leków 5) chęć zdobycia nowych leków
Stosunek lekarza do narkotyków: optymizm terapeutyczny (poleganie na lekach jako potężny składnik terapii), nihilizm terapeutyczny (odmowa przyjmowania nowych leków, przestrzeganie niektórych leków, nieufność do nowych leków)
Zgodność (przestrzeganie) pacjenta do leczenia 1) zrozumienie zaleceń lekarza i celów leczenia 2) dążenie do dokładnego przestrzegania zaleceń lekarza.
Obecnie na świecie jest około 100 000 leków, w Republice Białorusi zarejestrowanych jest ponad 4000, z czego około 300 to leki niezbędne. Badanie farmakologii pomaga nie utonąć w morzu narkotyków.
48. Podstawowe zasady leczenia i profilaktyki zatruć lekami. Terapia antidotum.
Klasyfikacja substancji toksycznych (OM):
1. Poprzez przynależność do pewnych klas związków chemicznych: barbiturany, benzodiazepiny, cyjanki.
2. Według pochodzenia: natura niebiologiczna (kwasy, zasady, sole metali ciężkich), toksyczne produkty odpadowe niektórych MB (toksyna botulinowa), pochodzenie roślinne (alkaloidy, glikozydy), pochodzenia zwierzęcego (jad węży i pszczół)
3. W zależności od stopnia toksyczności: a) skrajnie toksyczny (DL50< 1 мг/кг) б) высоко токсические (1-50) в) сильно токсические (50-500) г) умеренно токсические (500-5000) д) мало токсические (5000-15000) е) практически нетоксические (> 15.000)
4. Poprzez działanie toksykologiczne: a) paraliżujące nerwy (skurcz oskrzeli, duszenie) b) resorpcyjne skóry c) toksyczne ogólne (konwulsje hipoksyjne, śpiączka, paraliż) d) duszące e) łzowe i drażniące e) psychotropowe (upośledzona aktywność umysłowa, świadomość )
5. W zależności od obszaru preferencyjnego zastosowania: trucizny przemysłowe, pestycydy, trucizny domowe, bojowe środki chemiczne, substancje lecznicze.
6. W zależności od toksyczności leków: Lista A – leki, których przeznaczenie, stosowanie, dawkowanie i przechowywanie, ze względu na ich wysoką toksyczność, należy wykonywać z dużą ostrożnością. Ta sama lista obejmuje leki powodujące uzależnienie od narkotyków; lista B - leki, których mianowanie, stosowanie, dawkowanie i przechowywanie należy przeprowadzać z ostrożnością w związku z możliwymi powikłaniami podczas ich stosowania bez nadzoru lekarskiego.
Selektywnie toksyczne działanie leków.
A) kardiotoksyczne: glikozydy nasercowe, suplementy potasu, leki przeciwdepresyjne
B) neurotoksyczne: środki psychofarmakologiczne, oksychinoliny, aminoglikozydy
B) hepatotoksyczne: tetracykliny, chloramfenikol, erytromycyna, paracetamol
D) nefrotoksyczne: wankomycyna, aminoglikozydy, sulfonamidy
E) gastroenterotoksyczne: steroidowe leki przeciwzapalne, NLPZ, rezerpina
E) hematotoksyczne: cytostatyki, chloramfenikol, sulfonamidy, azotany, azotyny
G) pneumotoksyczne
Toksykokinetyka - bada wchłanianie, dystrybucję, metabolizm i wydalanie leków przyjmowanych w dawkach toksycznych.
Możliwe jest przedostanie się substancji toksycznych do organizmu a) dojelitowo b) pozajelitowo. Szybkość i kompletność wchłaniania odzwierciedla tempo rozwoju efektu toksycznego i jego nasilenie.
Dystrybucja w organizmie: Vd = D / Cmax - rzeczywista objętość, w której trująca substancja jest rozprowadzana w organizmie. Vd> 5-10 l/kg - OM trudno pozwolić na jego usunięcie (leki przeciwdepresyjne, fenotiazyny). Vd< 1 л/кг – ОВ легче удалить из организма (теофиллин, салицилаты, фенобарбитал).
Przedawkować- zmiany w procesach farmakokinetycznych: rozpuszczalność, związek z białkami, metabolizm ® znaczny wzrost wolnej frakcji leków ® działanie toksyczne.
Kinetyka pierwszego rzędu wraz ze wzrostem stężenia leku przekształca się w kinetykę rzędu zerowego.
Etap toksynogenny to terapia detoksykacyjna, etap somatogenny to terapia objawowa.
Toksydynamika . Główne mechanizmy działania toksycznego:
A) mediator: bezpośredni (według rodzaju blokady kompetycyjnej - FOS, psychomimetyki) i pośredni (aktywatory lub inhibitory enzymów)
B) interakcja z biocząsteczkami i strukturami wewnątrzkomórkowymi (substancje hemolityczne)
C) metabolizm według rodzaju letalnej syntezy (alkohol etylowy, tiofos)
D) enzymatyczny (jad węża itp.)
Rodzaje działania: lokalne, odruchowe, resorpcyjne.
Klasyfikacja zatruć:
1. Etiopatogenetyczne:
a) przypadkowe (samoleczenie, błędny odbiór)
b) umyślne (w celu popełnienia samobójstwa, zabójstwa, rozwinięcia w ofierze stanu bezradności)
2. Kliniczne:
a) w zależności od szybkości rozwoju zatrucia: ostre (przyjmowanie pojedynczej dawki lub w krótkim odstępie czasu toksycznej dawki substancji), podostre (opóźniony rozwój obrazu klinicznego po pojedynczej dawce), przewlekłe
b) w zależności od manifestacji głównego zespołu: uszkodzenie CVS, uszkodzenie DS itp.
c) w zależności od ciężkości stanu pacjenta: łagodny, umiarkowany, ciężki, skrajnie ciężki
3. Nozologiczny: uwzględnia nazwę leku, nazwę grupy substancji
Ogólny mechanizm śmierci w przypadku zatrucia:
A) porażka CVS:
1) obniżenie ciśnienia krwi, hipowolemia naczyń obwodowych, zapaść, brady - lub tachykardia (trójpierścieniowe leki przeciwdepresyjne, beta-blokery, blokery kanału wapniowego)
2) arytmie (częstoskurcz komorowy, migotanie – trójpierścieniowe leki przeciwdepresyjne, teofilina, amfetamina)
B) uszkodzenie ośrodkowego układu nerwowego: otępienie, śpiączka ® depresja oddechowa (leki, barbiturany, alkohol, leki nasenne)
C) drgawki, nadreaktywność i sztywność mięśni ® hipertermia, mioglobinuria, niewydolność nerek, hiperkaliemia
Triada toksykologiczna:
1) czas użytkowania, dawka i historia substancji ®.
2) ocena stanu świadomości według objawów: oddychanie, ciśnienie krwi, temperatura ciała body
3) dane laboratoryjne
Podstawowe zasady leczenia:
I. Pierwsza pomoc: sztuczne oddychanie, masaż serca, terapia przeciwwstrząsowa, kontrola równowagi wodno-elektrolitowej
II... Opóźnione wchłanianie i usuwanie niewchłoniętego OM z organizmu:
Cel: zakończenie kontaktu z OV
1. Droga pozajelitowa:
a) przez płuca:
1) zatrzymaj inhalację
2) substancje drażniące (amoniak, formaldehyd) ® w celu utrwalenia aktywnych ruchów, rozgrzania, podania tlenu i środków przeciwpieniących (w przypadku amoniaku środkiem przeciwpieniącym jest ocet, a w przypadku formaldehydu rozcieńczony roztwór amoniaku)
b) przez skórę: zmyć dużą ilością ciepłej wody z mydłem lub detergentem, swoistymi odtrutkami, zneutralizować i zakończyć narażenie na OM na skórze (FOS: przemyć wodą, usunąć 10-15% amoniakiem lub 5-6 % roztwór wodorowęglanu sodu z wodą; fenolkrezol: olej roślinny lub glikol etylenowy, ale nie olej wazelinowy, KMNO4: 0,5-1% roztwór kwasu askorbinowego lub równe objętości 3% roztworu nadtlenku wodoru i 3% roztworu kwasu octowego, CCl4, terpentyna, benzyna: ciepła woda z mydłem )
c) w przypadku wstrzyknięcia w kończynę: opaska uciskowa powyżej miejsca wstrzyknięcia
d) w przypadku kontaktu z oczami: płukać ciepłą solanką lub mlekiem przez 10-20 minut, zakroplić środek miejscowo znieczulający; w przypadku kontaktu z kwasami i zasadami nie można ich zneutralizować. Konieczna jest konsultacja z okulistą.
2. Droga dojelitowa: uwolnienie żołądka od OM, przyspieszenie pasażu,
a) usunięcie OM:
1) wstępne pobranie wody. Nie wolno spożywać mleka (z wyjątkiem żrących substancji trujących) i etanolu (z wyjątkiem metanolu).
2) wymioty – wskazane głównie w przypadku zatrucia dużymi tabletkami lub kapsułkami, które nie mogą przejść przez sondę. Może być wywołany odruchem lub wymiotami (NaCl: 1 łyżka stołowa na 1 szklankę wody; syrop Ipecac: dorośli 2 łyżki, dzieci 2 łyżeczki; musztarda: 1-2 łyżeczki na szklankę wody; apomorfina: 5-10 mg/kg podskórnie , z wyjątkiem dzieci poniżej 5 roku życia). Nie wywoływać wymiotów po spożyciu: rozpuszczalniki organiczne - niebezpieczeństwo wdychania, detergenty - substancje pieniące, drgawkowe - niebezpieczeństwo aspiracji, substancje żrące - uszkodzenie przełyku)
3) sonda do płukania żołądka - jest środkiem doraźnym i obowiązkowym. żołądek jest płukany, jeśli od zatrucia minęło nie więcej niż 4-6 godzin, czasem do 10 godzin; w przypadku zatrucia kwasem acetylosalicylowym – po 24 godzinach. Pacjent jest wstępnie zaintubowany rurką z nadmuchiwanym mankietem: w śpiączce przy braku kaszlu i odruchu krtaniowego. Żołądek myje się wodą lub roztworem soli o temperaturze 30 ° C, procedura trwa 4 godziny lub dłużej. Pod koniec prania węgiel aktywny i siarczan sodu.
b) zmniejszenie wchłaniania z przewodu pokarmowego: węgiel aktywowany wewnątrz po opróżnieniu żołądka + siarczan sodu lub magnezu. Cechy środków zmniejszających wchłanianie:
1) rozpuszczalniki organiczne: nie wywołują wymiotów, płukanie żołądka po intubacji, węgiel aktywny + parafina ciekła
2) detergenty: nie wywoływać wymiotów i wypłukiwać żołądka, należy podać dużo wody + środki przeciwpieniące (simetikon)
3) kwasy i zasady: nie wolno wywoływać wymiotów, jedynym wskazaniem do podania mleka jest płukanie żołądka przez rurkę nasmarowaną olejem roślinnym po podaniu narkotycznego środka przeciwbólowego. Do zatrucia kwasami - leki zobojętniające kwasy, do zatrucia zasadami - kwas cytrynowy lub octowy.
III... Usunięcie wchłoniętego OM z organizmu
a) wymuszona diureza (warunki: dostateczny przepływ krwi przez nerki i filtracja kłębuszkowa; wylać 20-25 litrów w ciągu 24 godzin)
b) hemodializa otrzewnowa
c) hemosorpcja
d) wymiana transfuzji krwi
e) wymuszona hiperwentylacja
IV... Terapia objawowa zaburzeń czynnościowych.
Antidota: 1) toksykotropowe – wiążące, neutralizujące i zapobiegające wchłanianiu OM: działające na zasadzie węgiel aktywowany działając na bazie chemicznej (unitol, penicylamina, pentacyna)
2) toksykokinetyczne - przyspieszają biotransformację OM (bromek trimedoksymu, tiosiarczan sodu, etanol, AO)
3) farmakologiczne - atropina, nalokson
4) odtrutki immunologiczne
Unitiol, sukcymer - wiąże metale ciężkie, metaloidy, glikozydy nasercowe. Esmolol wiąże teofilinę, kofeinę. Pentotan trójsodowy wapnia - tworzy kompleksy z metalami dwuwartościowymi i trójwartościowymi.
49. Receptura i jej struktura. Główne zasady wypisanie recepty. Państwowa regulacja zasad przepisywania i wydawania leków.
Przepis- jest to pisemny apel lekarza do farmaceuty z żądaniem wydania leku w pewna forma oraz dawkowanie wskazujące sposób jego użycia
W przepisie wyróżnia się następujące części:
1. Napis - tytuł, napis. Tu wpisuje się datę wystawienia recepty, nazwisko, inicjały i wiek pacjenta, nazwisko i inicjały lekarza.
2. Invocatio - kontakt z farmaceutą. Wyraża się go słowem „Przepis” (wziąć) lub skrótem (Rp.)
3. Designatio materiarum - oznaczenie lub nazwa produktów leczniczych ze wskazaniem ich dawek. W złożony przepis wykaz substancji leczniczych odbywa się w określonej kolejności. Pierwsza to główna substancja lecznicza (podstawa). Następnie pisze się adiuwany. Następnie wskazane są składniki korygujące smak, zapach, kolor leku (corrigens). Ostatnie do napisania są substancje, które nadają lekowi określoną postać dawkowania (składniki).
4. Subscriptio - recepta (wskazanie) do apteki. Wskazuje postać dawkowania, operacje farmaceutyczne wymagane do jego wytworzenia, liczbę wydanych dawek leku.
5. Sygnatura – poinstruowanie pacjenta, jak stosować lek.
6. Subscriptio medici - podpis lekarza, który wypisał receptę, jego imienna pieczęć.
Apel lekarza do farmaceuty, nazwa leków zawartych na recepcie, nazwa postać dawkowania a charakter operacji farmaceutycznych jest napisany w łacina... Nazwy leków, nazwy botaniczne roślin są pisane z Wielka litera... Recepta dla pacjenta jest wypisana w języku rosyjskim lub narodowym.
Ogólne zasady przepisywania:
1. Recepta wypisana jest na specjalnym formularzu, w zależności od przepisanego leku, wyraźnym pismem, tuszem lub długopis bez poprawek.
2. Recepta zawiera datę, miesiąc, rok, nazwisko, imię, nazwisko i wiek pacjenta, nazwisko, imię i nazwisko lekarza. Następnie pojawia się tekst przepisu, który wymienia nazwy substancji zawartych w przepisie w przypadku dopełniacza, wskazując ich ilość
3. Jednostką masy w recepturach jest gram lub JEDNOSTKA.
4. Przekroczenie maksymalnej dawki substancji trujących i silnych potwierdza się słownie
5. Recepta jest potwierdzona podpisem i imienną pieczęcią lekarza
W Republice Białorusi istnieje państwowa regulacja zasad przepisywania i wydawania leków.
50. Zasady przepisywania leków trujących, odurzających i silnie działających.
Lista A obejmuje leki, których wyznaczanie, stosowanie, dawkowanie i przechowywanie, ze względu na ich wysoką toksyczność, należy prowadzić z dużą ostrożnością. Ta sama lista obejmuje leki powodujące uzależnienie od narkotyków.
Lista B obejmuje leki, których wyznaczanie, stosowanie, dawkowanie i przechowywanie należy przeprowadzać ostrożnie w związku z możliwymi powikłaniami podczas ich stosowania bez nadzoru lekarskiego.
W przypadku trujących i silnych leków ustalono maksymalne wyższe dawki jednorazowe i dzienne. Te dawki są przeznaczone dla osób dorosłych powyżej 25 roku życia. Przy przeliczaniu dawek dla osób powyżej 60 roku życia, wrażliwość na wiek W celu różne grupy LS. Dawki leków hamujących ośrodkowy układ nerwowy, a także glikozydów nasercowych i diuretyków zmniejsza się o 50%, dawki innych trujących i silnych leków zmniejsza się do 2/3 dawki dla dorosłych. Dawki AB, sulfonamidów i witamin są zazwyczaj takie same dla wszystkich grupy wiekowe od 25 roku życia.
1. Środki odurzające (lista A) są przepisywane formularz recepty Forma 2. Jedna forma - jeden lek. Musi być: podpis i pieczęć lekarza prowadzącego, podpis ordynatora zakładu opieki zdrowotnej, okrągła pieczęć zakładu opieki zdrowotnej.
2. Leki trujące (lista A), silne (lista B) są przepisywane na formularzu recepty 1. Musi być podpis i pieczęć imienna lekarza, pieczęć placówki służby zdrowia.
51. Leki pod kontrolą. Leki na receptę.
Środki odurzające, trujące i silne są pod kontrolą (patrz sekcja 20)
A) Leki niezarejestrowane w Republice Białorusi i niedopuszczone do użytku służbowego
B) leki na wniosek pacjentów i ich bliskich bez badania pacjenta i ustalenia diagnozy
C) recepty na środki odurzające do wstrzykiwań, eter znieczulający, chloroetyl, pentamina, fluorotan, oksymaślan sodu w ampułkach, oksymaślan litu, siarczan baru do fluoroskopii.
52. Modele farmakokinetyczne (jednokomorowe i dwukomorowe), ilościowe prawa wchłaniania i eliminacji leków.
![]() Model jednokomorowy.
Model jednokomorowy.
Cały organizm jest jednym, jednorodnym pojemnikiem. Założenia:
1) między zawartością leku w krwiobiegu a jego stężeniem w tkankach pozanaczyniowych ustala się szybki dynamiczny rozwój
2) Lek jest szybko i równomiernie rozprowadzany w całej objętości krwi
3) Eliminacja leków podlega kinetyce pierwszego rzędu: tempo spadku zawartości leku we krwi jest proporcjonalne do jego stężenia
Jeśli mechanizmy eliminacji leku (biotransformacja w wątrobie, wydzielanie nerkowe) nie zostaną nasycone wprowadzeniem dawki terapeutycznej, zostanie wyświetlony logarytmiczny wykres zmian stężenia w osoczu w czasie liniowy.
Skłonić Oś lognormalna - Kel, gdzie Kel jest stałą szybkości eliminacji i ma wymiar czasu-1. Wartość C0 uzyskuje się poprzez ekstrapolację wykresu na przecięcie z osią rzędnych. Stężenie leku w osoczu(Ct) w dowolnym momencie t po podaniu do organizmu wynosi:
Ln Ct = Ln C0 - kt. Stała eliminacji Kel, Vd i całkowity klirens (CL) są powiązane wyrażeniem: CL = k × Vd
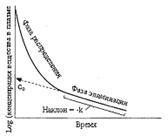 Model dwukomorowy.
Model dwukomorowy.
Często po dostaniu się leku do organizmu nie jest możliwe szybkie osiągnięcie równowagi między zawartością leku we krwi a jego stężeniem w płynie pozanaczyniowym. Następnie uważa się, że w agregacie tkanek i płynów biologicznych organizmu można wyróżnić dwie komory, które różnią się stopniem dostępności do penetracji leków. Komora centralna zawiera krew (często z intensywnie ukrwionymi narządami - wątroba, nerki), komora obwodowa - płyn śródmiąższowy narządy wewnętrzne i tkaniny.
Otrzymany wykres pokazuje początkowy Faza dystrybucji ( Czas potrzebny do osiągnięcia przez lek stanu równowagi między komorą centralną i obwodową a po niej powolnym Faza eliminacji Pierwsze zlecenie.
Wartość C0, uzyskana przez ekstrapolację Fazy eliminacji przed przekroczeniem rzędnej. C0 służy do obliczania objętości dystrybucji i stałej eliminacji. Wzory do obliczania Ct i Cl podane dla modelu jednokomorowego są również stosowane w fazie eliminacji leków spełniających warunki modelu dwukomorowego.
53. Selektywność i specyficzność działania leków. Terapeutyczne, uboczne i toksyczne działanie leków, ich istota z punktu widzenia koncepcji receptorów. Strategia terapeutyczna zwalczania skutków ubocznych i toksycznych leków.
Specyficzność- wtedy lek wiąże się z rodzajem receptora, który jest dla niego ściśle specyficzny.
Selektywność- ma to miejsce, gdy lek jest w stanie wiązać się z jednym lub kilkoma rodzajami receptorów dokładniej niż inne.
Bardziej korzystnie jest używać terminu selektywność, ponieważ jest mało prawdopodobne, aby jakakolwiek cząsteczka leku mogła wiązać się tylko z jednym typem cząsteczki receptora, ponieważ liczba potencjalnych receptorów u każdego pacjenta ma znaczenie astronomiczne.
Działanie terapeutyczne- główny pożądany efekt farmakologiczny oczekiwany od danego preparatu farmakologicznego.
Skutki uboczne- te efekty, które występują, gdy substancje są stosowane w dawkach terapeutycznych i stanowią spektrum ich działania farmakologicznego.
Efekty toksyczne- działania niepożądane objawiające się tym lekiem, gdy opuszcza on zakres terapeutyczny.
Zależności między działaniem terapeutycznym a toksycznym leków na podstawie analizy mechanizmów receptorowo-efektorowych:
1) działanie terapeutyczne i toksyczne, w którym pośredniczy ten sam mechanizm receptorowo-efektorowy (prazosyna działa jako alfa-selektywny antagonista na naczyniowe receptory SMC i ma działanie hipotensyjne w nadciśnieniu pierwotnym, ale przy dużych dawkach pacjent może doświadczać niedociśnienia ortostatycznego)
2) działanie terapeutyczne i toksyczne, w którym pośredniczą identyczne receptory, ale różne tkanki lub różne drogi efektorowe (glikozydy nasercowe są stosowane w celu zwiększenia kurczliwości mięśnia sercowego, jednocześnie zaburzają funkcję przewodu pokarmowego, widzenie z powodu blokady Na + / K + -ATPaza błony komórkowej)
3) działanie terapeutyczne i toksyczne, w którym pośredniczą różne typy receptorów (na przykład noradrenalina ma działanie nadciśnieniowe przez a1-Ap, ale jednocześnie powoduje tachykardię przez b1-Ap)
Strategia terapeutyczna w walce z terapeutycznymi i ubocznymi skutkami leków:
1. Lek należy zawsze podawać w najmniejszej dawce, która powoduje akceptowalny efekt terapeutyczny.
2. Zmniejszenie dawki jednego leku z powodu wyznaczenia innego leku o podobnym działaniu, ale przez różne receptory i o innym profilu toksyczności.
3. Selektywność działania leków można zwiększyć poprzez kontrolowanie stężenia leków w rejonie receptorów różnych części ciała (miejscowe stosowanie leków – wziewne stosowanie salbutamolu w astmie oskrzelowej)