Effet pharmacologique. Mécanismes physico-chimiques et chimiques de l'action des substances médicinales. Stratégie de thérapie médicinale individuelle
Blocator H 1-gismine récepteurs de la première génération. L'action sur le système nerveux central est due aux récepteurs cérébraux H 3-pistamiques de blocage et à l'oppression des structures cholinergiques centrales. Supprime le spasme d'une musculature lisse (action directe), réduit la perméabilité des capillaires, avertit et affaiblit les réactions allergiques, a un effet local particulier, anti-cicatrisant et de sédation, bloque modérément les cholinorécepteurs de ganglions végétatifs, a une action de la pilule endormie.
Témoignage - réactions anaphylactiques et anaphylactoïdes (en thérapie complexe); - gonflement du quinque;
Maladie sérique; - d'autres conditions allergiques actives (en thérapie complexe et dans les cas où l'utilisation de la forme sur table est impossible).
Effets secondaires
Du côté système nerveux: somnolence, faiblesse, réduction du taux de réaction psychomotrice, troubles de la coordination des mouvements, vertiges, tremblements, irritabilité, euphorie, excitation (surtout chez les enfants), insomnie.
Du côté système respiratoire: sécheresse de la muqueuse orale, nez, bronchi (augmentation de la viscosité humide).
De la part des organes de formation de sang: anémie hémolytique, thrombocytopénie, agranulocytose.
Du côté du système cardio-vasculaire: Pression artérielle réduite, tachycardie, extrasystole.
Du système urinaire: dépréciation de la miction.
Réactions allergiques: urticaire, photosensibilité, éruption cutanée, démangeaisons.
Loratadine (Claritin)
effet pharmacologique
Préparation antiallergique, blocage sélectif de l'histamine périphérique H 1 -Receurs. La loratadine est une connexion tricyclique avec un effet antihistaminique prononcé. Il a un effet antiallergique rapide et à long terme.
La loratadine ne pénètre pas dans la Colombie-Britannique et n'affecte pas le CNS. N'a pas d'action anticholinerinergique ou sédative cliniquement significative, c'est-à-dire Ne provoque pas de somnolence et n'affecte pas le taux de réactions psychomotrices lorsqu'il est appliqué dans les doses recommandées. La réception de la claritine de la drogue ne prolonge pas l'intervalle QT par ECG. Pour traitement long Non observé cliniquement changements significatifs Indicateurs de fonctions vitales, données d'inspection physique, résultats de la recherche en laboratoire ou ECG.
La loratadine n'a pas de sélectivité significative par rapport aux récepteurs de l'histamine H 2. N'inhibe pas l'adhérence inverse de la norépinéphrine et n'affecte pratiquement pas le système cardiovasculaire ni la fonction du pilote de rythme
Effets secondaires
Du système nerveux: Chez les enfants âgés de 2 à 12 ans - maux de tête (2,7%), nervosité (2,3%), fatigue (1%); chez les adultes - mal de crâne (0,6%), somnolence (1,2%), insomnie (0,1%).
Chez les adultes - une augmentation de l'appétit (0,5%).
En période de post-marketing
Du système nerveux: rarement (< 1/10 000) - головокружение, утомляемость.
Du côté système digestif: rarement (< 1/10 000) - сухость во рту, желудочно-кишечные расстройства (тошнота, гастрит), нарушение функции печени.
Réactions allergiques: rarement (< 1/10 000) - сыпь, анафилаксия.
Du côté du système cardiovasculaire: rarement (< 1/10 000) - сердцебиение, тахикардия.
Du côté peau pokrov: rarement (< 1/10 000) - алопеция.
Les indications
Saison (pollinose) et rhinite allergique toute l'année et conjonctivite allergique (pour éliminer les symptômes associés à ces maladies - éternuement, démangeaisons la membrane muqueuse du nez, rhinores, sensations de brûlure et de démangeaisons dans les yeux, de la déchirure); - urticaire idiopathique chronique; - Maladies cutanées d'origine allergique.
33. signifie éliminer les manifestations générales réactions allergiques Tapez le choc anaphylactique. Epinéphrine, Uefillin, Prednisiralon. Les principaux effets pharmacologiques, le but de la nomination de chaque médicament et des effets indésirables.
Antiallergique signifie (Antiallergica; péché. Agents désensibilisants) - Drogues Avertissement ou affaiblissement des manifestations de réactions allergiques. Un des chemins possibles La prévention et le traitement des réactions allergiques est la méthode ainsi appelée. Hyposensibilisation spécifique, c'est-à-dire baisser la sensibilité du corps à tout antigène en administrant à plusieurs reprises l'antigène lui-même à de faibles doses qui ne causent pas de manifestations d'allergies. Dans ce cas, le corps perd progressivement la sensibilité à l'antigène administré.
Adrénaline
L'effet pharmacologique de l'adrénomimétique a un effet stimulant directement sur les α et les orrénorécepteurs. Sous l'action de l'épinéphrine (adrénaline), due à la stimulation des α-adrénorécepteurs, une augmentation de la teneur en calcium intracellulaire dans des muscles lisses se produit. Augmente l'activité de la phospholipase C (à travers la stimulation de la protéine G) et de la formation d'inositatriphosphate et de diacylglycérol. Il a une action prononcée sur le système cardiovasculaire. Augmente la fréquence et la force des abréviations cardiaques, des chocs et des minuscules volumes cardiaques. Améliore la conduction AV, améliore l'automatisme. Augmente la nécessité de myocardium dans l'oxygène. Le rétrécissement des navires des organes de la cavité abdominale, du cuir, de la membrane muqueuse, dans une moindre mesure - des muscles squelettiques. Augmente la pression artérielle (principalement systolique), l'épinéphrine (adrénaline) détend les muscles lisses de la bronche, abaisse la moto gastro-intestinale de ton et de moto, élargit les élèves, contribue à une diminution de la pression intra-oculaire. Provoque une hyperglycémie et augmente la teneur dans le plasma d'acides gras libres. Indications Réactions allergiques du type immédiat (y compris l'urticaire, le choc angoidichique, le choc anaphylactique), le développement de médicaments, sérums, transfusion sanguine, utilisation alimentaire, mord d'insectes ou introduction d'autres allergènes. Asthme bronchique (attaque de liaison), bronchospasme lors de l'anesthésie. Afin d'arrêter sa saignement. Effet secondaire sur le côté du système cardiovasculaire: angine, bradycardie ou tachycardie, battement de coeur, augmentation ou diminution de la pression artérielle; Lorsqu'il est utilisé dans des doses élevées - des arythmies ventriculaires; Rarement - Arythmie, douleur thoracique. Du système nerveux: maux de tête, état anxieux, tremblements, vertiges, nervosité, fatigue, troubles psychonurotiques
Eufillin
effet pharmacologique
Agent broncholitiquement, dérivé de xanthine; inhibe la phosphodiestérase, augmente l'accumulation de monophosphate d'adénosine cyclique dans les tissus, bloque les récepteurs de l'adénosine (purine); Réduit le flux d'ions calcium à travers des cellules des membranes cellulaires, réduit l'activité contractile des muscles lisses. Relaxant les muscles de la bronche, augmente le jeu de mucociliarie, stimule la réduction de la diaphragme, améliore la fonction des muscles respiratoires et intercostaux, stimule le centre respiratoire, augmente sa sensibilité au gaz de dioxyde de carbone et améliore la ventilation alvéolaire, qui entraîne finalement une diminution de la la gravité et la fréquence des épisodes d'apnée. Normaliser la fonction respiratoire contribue à la saturation du sang avec de l'oxygène et une diminution de la concentration en dioxyde de carbone. Il a un effet stimulant sur l'activité du cœur, augmente la force et le nombre d'abréviations cardiaques, augmente le flux sanguin coronaire et la nécessité de myocardium dans l'oxygène. Réduit le ton des vaisseaux sanguins (principalement des navires du cerveau, du cuir et du rein). Il a un effet de ventilation périphérique, réduit la résistance vasculaire pulmonaire, réduit la pression dans le "petit" cercle de cercle. Augmente le flux sanguin rénal, a un effet diurétique modéré. Élargit des gales extrahépatiques. À travers l'agrégation plaquettaire (supprime le facteur d'activation des plaquettes et PGE2 alpha), augmente la stabilité des érythrocytes à la déformation (améliore les propriétés rhéologiques du sang), réduit la thrombose et normalise la microcirculation. Il a un effet tocolique, augmente l'acidité du jus de gastrie. Lorsqu'il est utilisé dans de grandes doses, il a un effet énylectogène.
Effets secondaires
Du système nerveux: vertiges, maux de tête, insomnie, excitation, anxiété, irritabilité, tremblements.
Du côté du système cardiovasculaire: terrain de coeur, tachycardie (y compris le fœtus lors de la grossesse au troisième trimestre), les arythmies, la cardialie, la diminution de la pression artérielle, augmentent la fréquence des attaques d'angandes.
Du système digestif: Gastralgie, nausée, vomissements, reflux gastro-oesophagien, brûlures d'estomac, aggravation de la maladie ulcéreuse, diarrhée, avec réception à long terme - diminution de l'appétit.
Réactions allergiques: Éruption cutanée, démangeaisons, fièvre.
Autres: douleur mammaire, Tachipne, sentiment de marées à faire face, albuminurie, hématurie, hypoglycémie, renforcement de la diurèse, transpiration accrue.
Les indications
Syndrome de broncho-constructif de toute origine: asthme bronchique (sélection de médicaments chez les patients atteints de stress physique de l'asthme et à un remède supplémentaire pour d'autres formas), maladie pulmonaire obstructive chronique, emphysème pulmonaire, bronchite obstructive chronique, hypertension pulmonaire, coeur "pulmonaire", nuit apnée.
Prednisolone
effet pharmacologique
GK synthétiques. Il a une action anti-inflammatoire prononcée. Le médicament freine le développement des symptômes de l'inflammation. L'accumulation de macrophages, de leucocytes et d'autres cellules dans la zone d'inflammation. Inhibe la phagocytose, la libération d'enzymes microsomales, ainsi que la synthèse et la libération de médiateurs d'inflammation. Provoque une diminution de la perméabilité des capillaires, la migration de freinage des leucocytes.
Améliore la synthèse de la lipomoduoduline, l'inhibiteur de la phospholipase A2, qui libère de l'acide arachidonique à partir de membranes phospholipides avec une inhibition simultanée de sa synthèse.
Le mécanisme d'action immunosuppressive de la prednisolone n'est pas entièrement étudié. Le médicament réduit la quantité de t-lymphocytes, de monocytes et de granulocytes acidophiliques, ainsi que de la liaison d'immunoglobulines avec des récepteurs sur la surface de la cellule, inhibe la synthèse ou la libération d'interleukines en réduisant la blastogenèse de t-lymphocytes; Réduit la réponse immunologique précoce. La pénétration des complexes immunologiques à travers les membranes ralentit également et réduit la concentration de composants et d'immunoglobulines complémentaires.
Prednisolone agit sur la partie distale des tubules rénaux, améliorant l'aspiration inverse de sodium et d'eau, ainsi qu'une augmentation de la libération d'ions de potassium et d'hydrogène.
Prednisolone inhibe la sécrétion de la glande pituitaire ACTH, ce qui entraîne une diminution de la production de corticostéroïdes et d'androgènes de croûtes surrénales. Après une longue utilisation du médicament à fortes doses, la fonction surrénale peut être restaurée au cours de l'année et, dans certains cas, la suppression résistante de leur fonction se développe. Prednisolone améliore le catabolisme des protéines et induit des enzymes impliquées dans le métabolisme des acides aminés. Il ralentit la synthèse et améliore le catabolisme des protéines dans les tissus musculaires lymphatiques, connectifs et musculaires. Avec une utilisation prolongée, l'atrophie de ces tissus est possible (ainsi que la peau).
Augmente la concentration en glycémie par induction d'enzymes de la gluchegénèse dans le foie, la stimulation du catabolisme des protéines (qui augmente la quantité d'acides aminés pour la glyconèse) et réduire la consommation de glucose dans les tissus périphériques. Cela conduit à l'accumulation de glycogène dans le foie, une augmentation de la concentration en glycémie et une augmentation de la résistance à l'insuline.
Les indications
Maladies endocrinologiques:
Manque de cortex surrénalien: primaire (maladie de Addison) et secondaire; - syndrome adrénénitaire (hyperplasie congénitale des glandes surrénales); - absence aiguë de cortex surrénalien;
Devant les interventions chirurgicales et dans des maladies sévères et des blessures chez les patients atteints de défaillance surrénalienne; - Thyroïdite subaiguë.
Maladies allergiques lourdes résistantes à une autre thérapie: - Dermatite de contact; - la dermatite atopique; - maladie du sérum; - réactions d'une sensibilité accrue à la drogue;
Permanent ou saisonnier rhinite allergique; - Réaction anaphylactique; - Edema Angioedema.
Maladies rhumatismes:
Arthrite rhumatoïde, arthrite rhumatoïde juvénile (en cas de résistance aux autres méthodes de traitement);
Maladies dermatologiques: - dermatite exfoliative; - dermatite bulleuse de la forme de forme;
Dermatite sévère séborrhéique; - érythème multiforme grave (Stevens-Johnson Syndrome);
Effets secondaires
Avec l'utilisation à court terme de la prednisolone (ainsi que d'autres GKS), les effets secondaires sont rarement observés. Lors de l'application de la prednisone, les effets secondaires suivants sont possibles pendant une longue période.
De l'équilibre d'eau et d'électrolyte: Retard dans l'organisme de sodium et liquide, hypokaliémie.
Sur le côté du système musculo-squelettique: Faiblesse musculaire, myopathie de stéroïdes, perte de masse musculaire, ostéoporose, fracture de la colonne vertébrale compression.
Du système digestif: Ulcer stéroïde avec éventuelle ardoise et saignement, pancréatite, météorisme, œsophagite ulcéreuse, trouble digestif, nausée, appétit accru.
Réactions dermatologiques: Atrophie de la peau, Stria, acné, cicatrisation lente de la plaie, amincissement de la peau, petechie, hématome, érythème, transpiration accrue, dermatite allergique, urticaire, œdème d'Œdema.
Du SNC et du système nerveux périphérique: Une pression intracrânienne accrue avec un syndrome de mamelon stagnant d'un nerf visuel (survient le plus souvent chez les enfants, après une réduction rapide de la dose, des symptômes - maux de tête, une détérioration de l'acuité visuelle, des biais dans les yeux); Crampes, vertiges, maux de tête, troubles du sommeil.
De l'état endocrinien: Insuffisance secondaire sur l'adrénale et hypothalamique-pituitaire (surtout lors de situations stressantes: maladie, blessure, chirurgie); Syndrome de Cushing.
Autres: réactions anaphylactiques, réactions d'hypersensibilité; Supposons que l'artérite, une augmentation du poids corporel, évanouissement.
1. L'essence de la pharmacologie en tant que science. Sections et zones de pharmacologie moderne. Les principaux termes et concepts de pharmacologie sont l'activité pharmacologique, l'action, l'efficacité substances chimiques.
Pharmacologie - Science des médicaments dans tous les aspects - fondation théorique Thérapies:
a) Science sur l'interaction des produits chimiques avec des systèmes vivants
b) Science sur la gestion des processus de l'activité vitale du corps à l'aide de produits chimiques
Développement de la pharmacologieil va dans deux directions principales: recherche fondamentale Clarifier les principes et les mécanismes de l'action de la LS et du développement drogues efficaces Comme base pour le traitement des maladies.
La pharmacologie est divisée en:
1. Général- étudie les modèles généraux d'interaction substances médicinales Avec des organismes vivants.
Privé- considère que des groupes pharmacologiques et des individus spécifiques
2. Pharmacologie expérimentale (basique) - étudie l'effet des médicaments dans l'expérience.
Pharmacologie clinique - Études Efficacité clinique et sécurité
les applications de médicaments chez les patients optimise le programme de traitement du patient en ce qui concerne
ses états.
Toxicologie - Études Effet toxique sur les organes diverses substances (comprenant
et médicinal).
Sections de pharmacologie moderne:
1) pharmacodynamique - étudier a) l'impact des médicaments sur le corps humain, b) l'interaction de divers médicaments dans le corps tout en les nominant simultanément, c) l'effet de l'âge et diverses maladies sur l'action de la LS.
2) pharmacocinétique - étudie l'aspiration, la distribution, le métabolisme et l'excrétion des médicaments (c'est-à-dire que le corps d'un patient réagit à LS)
3) pharmacogénétique- étudie le rôle des facteurs génétiques dans la formation de la réponse pharmacologique du corps sur le réseau local
4) pharmacoéconomique- évalue les résultats de l'utilisation et du coût des médicaments pour prendre une décision sur l'application pratique ultérieure.
5) pharmacoépideyologie - étudie l'utilisation de drogues et de leurs effets au niveau des populations ou grands groupes les gens pour assurer l'utilisation des ls les plus efficaces et les plus sûrs
Termes majeurs et concepts:
Activité pharmacologique (biologique) - la propriété de la substance pour causer des changements dans le biosystème (corps humain). Substances pharmacologiques \u003d biologiquement substances actives (BAV)
effet pharmacologique - l'effet des médicaments sur l'objet et sa cible
Effet pharmacologique - résultat de l'action de la substance dans le corps (modification des processus physiologiques, biochimiques, structures morphologiques) - quantitatif, mais pas changement de qualité dans un état de biosystèmes (cellules, tissus, organes).
Efficacité de LS. - la capacité du réseau local à appeler certains nécessaires dans ce cas Effets pharmacologiques dans le corps. Il est estimé sur la base de «preuves substantielles» - des études et des essais cliniques bien contrôlés adéquats effectués par des experts avec une formation scientifique et une expérience scientifique pertinentes pour étudier les médicaments de ce type (FDA)
2. Sources et étapes de la création de médicaments. Médicaments - Generics, placebo - Effets définissant les concepts de la substance médicamenteuse, de la médecine, de la dose et de la posologie.
Sources de création de médicaments:
a) Matières premières naturelles: plantes, animaux, minéraux, productivité Produits de micro-organismes (glycosides coeur, insuline de porc, ab)
b) BAV naturel modifié
c) Produits de synthèse chimique (méthodes: criblage pharmacologique, conception moléculaire, reproduction d'amines biogéniques, modification ciblée de molécules avec activité déjà connue, synthèse de métabolites pharmacologiquement actifs, découvertes aléatoires (méthode "Serenity"))
d) Produits d'ingénierie génétique (insuline recombinante, interférons)
Étapes de la création de médicaments:
1. Synthèse de LS dans le laboratoire chimique
2. Évaluation préclinique de l'activité et des effets indésirables du ministère de la Santé et des autres. Organisations
3. Essais cliniques de l'examen de la documentation Le Comité pharmacologique est effectué après avoir terminé chaque phase. Le médicament peut être retiré à n'importe quel stade. (I PHASE - Évaluation de la tolérance sur des volontaires sains de 20 à 25 ans, II - sur des patients atteints de bénévolat moins de 100 personnes souffrant d'une maladie de certaines maladies, phase III - des études cliniques multicentrales dans de grands groupes de personnes (jusqu'à 1000 personnes ), phase IV - Surveillance du médicament 5 ans après sa permission officielle (effectuée sur un grand nombre de patients (au moins 1 000 à 5 000 personnes). Après la fin de la phase III des essais cliniques, la documentation est enregistrée à nouveau à la Comité pharmacologique (le dossier total peut comporter jusqu'à 1 million de pages) et dans les 1-2 ans est enregistré dans le registre des médicaments et des produits de l'État. prescription médicale. Seulement après que la préoccupation pharmacologique a le droit de démarrer une libération industrielle drogue Et sa distribution via le réseau de pharmacie.
Le médicament générique est un médicament non indentable, qui est la perception du médicament initial, sur la substance active dont la période de protection des brevets a expiré. Peut différer de la drogue d'origine dans la composition des substances auxiliaires. L'exigence requise pour la vente de génériques est la preuve d'équivalence pharmaceutique, biologique et thérapeutique de la matière première. Les médicaments génériques sont toujours moins chers que leurs homologues de marque, car La société ne dépensait pas de fonds pour 10-15 médicaments d'études d'été, mais utilise des données prêtes à l'emploi.
Placebo - Toute composante thérapie qui n'a pas d'impact biologique spécifique sur la maladie, qui fait l'objet de traitement.
Appliqué au contrôle lors de l'évaluation de l'action des médicaments et afin de bénéficier au patient sans aucun moyens pharmacologiques En conséquence, seul impact psychologique (c'est-à-dire effet placebo).
Tous les types de traitement ont composant psychologiqueou livrer satisfaction ( effet placebo), Soit anxieux (effet nocebo). Exemple d'effet placebo: amélioration rapide chez le patient infection virale Lors de l'utilisation d'antibiotiques. L'effet placebo favorable est lié à influence psychologique sur le patient. Ce sera le maximum que lorsqu'il sera utilisé en combinaison avec des méthodes de traitementavoir un effet spécifique prononcé. Substances coûteuses Comme un placebo contribue également à la réalisation d'une plus grande réponse.
Indication pour placebo:
1) faible violation mentale
2) soutien psychologique Patient avec incurable maladie chronique ou soupçonné un diagnostic lourd
Médicament - toute substance ou produit utilisé pour modifier ou explorer des systèmes physiologiques ou des conditions pathologiques pour la prestation des prestations (OMS, 1966); Substances individuelles, mélanges de substances ou composition de la composition inconnue, qui ont des propriétés thérapeutiques éprouvées.
Substance médicinale - Composé chimique individuel utilisé comme médicament.
Formulaire de dosage - confortable pour application pratique La forme attachée au médicament pour obtenir l'effet thérapeutique ou préventif nécessaire.
Traitement médical - médicament à une certaine formulaire de dosage, autorisé par le gouvernement.
Par exemple: la substance médicinale est un antibiotique d'ampicilline, un trihydrate de dosage de l'ampicilline, qui peut être produit sous forme de comprimés ou de gélules. Le médicament est la pilule du trihydrate d'ampicilline de 0,25 g.
Façons d'introduire des médicaments dans le corps et leurs caractéristiques. Élimination de la pression des médicaments.
mais. Voie d'administration entérale: oralement, sublingual, transbuccitale, rectale, à travers la sonde b. Route parentérale de l'administration: intraveineuse, sous-cutanée, intramusculaire, ... 2. Pour une exposition locale: capturée (épicataire), sur des muqueuses, dans la cavité (abdominale, pléurale, articulation), en tissu ...Transfert de médicaments par des barrières biologiques et ses variétés. Les principaux facteurs affectant le transfert de médicaments dans le corps.
1) Filtration (diffusion aqueuse) - mouvement passive des molécules de la substance en fonction du gradient de concentration à travers les pores remplis dans la membrane de chacune ... 2) Diffusion passive (diffusion lipidique) - le mécanisme principal du transfert de LV , ... 3) Transport avec des transporteurs spécifiques - Transfert LV avec intégré dans la membrane des transporteurs (plus souvent ...Transférer par des membranes de substances médicinales à ionisation variable (l'équation d'ionisation de Gendar Gasselbalch). Principes de contrôle de transfert.
Tous les LS sont des acides faibles ou des terrains faibles ayant leurs significations de la constante d'ionisation (RK). Si la valeur polaire du milieu est égale à la valeur du médicament RK, 50% de ses molécules seront sous ionisées et 50% dans l'état non ionisé et le milieu du médicament sera neutre.
DANS un environnement acide (pH moins que le RK), où il y a un excès de protons, l'acide faible sera de la forme indiscible (R-Cooh), c'est-à-dire Sera associé à un protoné protoné. Une telle forme acide est non chargée et bien soluble dans les lipides. Si le pH est décalcuté sur le côté alcalin (c'est-à-dire que le pH deviendra plus de la RK), l'acide commencera ensuite à dissocier et à perdre le proton, tout en se transformant en une forme déverrouillée, qui a une charge et est mal soluble dans les lipides.
Dans un milieu alcalin, où il y a un déficit de protons, la base faible sera sous la forme indiscible (R-NH 2), c'est-à-dire Il sera non passué et privé de charge. Cette forme de base est bien soluble dans les lipides et est rapidement absorbée. Dans le milieu acide, il y a un excès de protons et une base faible commencera à dissocier, à lier les protons et à former la forme de base chargée protonée. Une telle forme est mal soluble dans les lipides et est mal absorbée.
D'où, l'absorption des acides faibles se déroule principalement dans un environnement acide et des bases faibles en alcaline.
Caractéristiques du métabolisme des acides faibles (SC):
1) Estomac: SK dans la teneur en acide de l'estomac est non ionisée et dans un milieu alcalin de l'intestin grêle, il sera dissocié et les molécules SC acquiert une charge. Par conséquent, l'absorption des acides faibles sera la plus intense de l'estomac.
2) Dans le sang, le milieu est plutôt alcalin et les molécules SK Skidle vont se passer sous forme ionisée. Le filtre du glomère rénal passe à la fois des molécules ionisées et non ionisées, donc malgré la charge de la molécule, le SC sera émis dans urine primaire
3) Si l'urine est alcaline, l'acide restera sous forme ionisée, ne sera pas capable de les réabsorber dans le sang et de se démarquer de l'urine; L'urine est acide, le médicament passe à une forme non ionisée, qui est facilement réactivée dans le sang.
Caractéristiques du métabolisme des terrains faibles: contraire de manière opposée (l'absorption est meilleure dans les intestins; dans l'urine alcaline soumise à la réabsorption)
Donc pour accélérer l'élimination du corps de la faible urine acide doit être nettoyée et d'accélérer l'élimination d'une base faible doit être acidifiée (désintoxication par POPOV).
La dépendance quantitative du processus d'ionisation du médicament avec un pH différent du support vous permet d'obtenir l'équation Henderson-Hasselbach.:
Où PKA correspond à la valeur du pH dans laquelle la concentration de formes ionisées et non ionisées est en équilibre .
L'équation de Gasselbach nous permet d'estimer le degré d'ionisation des médicaments à une valeur de pH donnée et de prédire la probabilité de sa pénétration à travers la membrane cellulaire.
(1)Pour diluer acide, a,
HA ↔ H + + A - L'HA est une concentration de formes non ionisées (protonées) de l'acide et une - - la concentration de forme ionisée (non compromée).
(2) pour base faible, b,
BH + ↔ H + + B, où BH + est la concentration de la forme de base protonée, B est une concentration de forme non déplacée
Connaissant le pH du milieu et la substance RCA, il est possible de déterminer le degré d'ionisation du médicament en fonction du logarithme calculé, et donc le degré de son aspiration de tube digestif, réabsorption ou excrétion par les reins quand valeurs différentes pH urine et ainsi de suite.
Principes de contrôle de transfert.
Accélérer l'aspiration
Ø d'acides faibles (ASC) - le pH du jus de gastrie devrait être acide;
Ø La base faible - le pH du jus gastrique devrait être neutre.
Accélérer l'élimination
Ø d'acides faibles - urine coincée;
Ø Bases faibles - Urine Acidifier.
Transfert de médicaments dans le corps. Diffusion de l'eau et diffusion dans les lipides (loi FIC). Transport actif.
Le transfert de LS dans le corps peut être effectué avec de l'eau et de la diffusion lipidique, du transport actif, de l'endo et de la pinocytose.
Caractéristiques Transfert LS dans le corps de la diffusion de l'eau:
1. Couvertures épithéliales (Duals muqueux, cavité buccale, etc.) - Diffusion de l'eau de seules de très petites molécules (méthanol, lithium ions, etc.)
2. Capillaires (sauf pour le cerveau) - filtrage des substances avec du poids moléculaire jusqu'à 20-30 mille oui.
3. Capillaires cérébraux - N'a principalement pas de pores d'eau, à l'exception des zones d'hypophyse, d'épiphyse, de zones de ventriculaire, de plexus choroïdien, d'altitude médiane
4. Placenta - n'a pas de AUMA (bien que la question soit controversée).
5. La liaison des médicaments avec des protéines sanguines empêche leur sortie du sang, et donc la diffusion de l'eau
6. La diffusion dans l'eau dépend de la taille des molécules LS et des pores d'eau
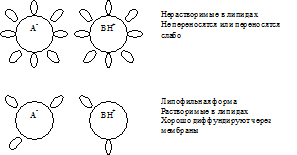
Caractéristiques de la diffusion de lipides:
1. Le mécanisme principal de LS transfère à travers les membranes cellulaires
2. La lipophilicité de la substance diffusable est déterminée (c'est-à-dire le coefficient de distribution d'huile / eau) et le gradient de concentration peut être limité à une très faible solubilité de la substance dans l'eau (qui empêche la pénétration de médicaments dans la phase aqueuse des membranes )
3. Connexions non polaires facilement diffuses, ions difficiles.
Toute diffusion (et eau, dans les lipides) obéit la loi de la diffusion fika:
Taux de diffusion - la quantité de molécules de médicaments portables par unité de temps; C 1 est la concentration de la substance à l'extérieur de la membrane; C 2 est la concentration de la substance de l'intérieur de la membrane.
Conséquence de la loi fika:
1) la filtration LS est plus élevée que sa concentration sur le site d'injection (s absorbée de la surface dans l'intestin plus que dans l'estomac, l'absorption des médicaments dans l'intestin est plus rapide)
2) Le filtrage des faibles est supérieur à la plus grande que la concentration de médicaments sur le site d'injection
3) La filtration LS est supérieure à l'épaisseur la moins épaisseur de la vaste membrane biologique (l'épaisseur de la barrière dans les alvéoles des poumons est nettement inférieure à la peau, de sorte que la vitesse d'absorption est plus élevée dans les poumons)
Transport actif - Transférer LC indépendamment du gradient de concentration en utilisant aTF EnergyIl est caractéristique des molécules polaires hydrophiles, un certain nombre d'ions inorganiques, de sucres, d'acides aminés, de pyrimidines.
Caractérisé: a) Sélectivité à certains composés b) La possibilité de concurrence de deux substances pour un mécanisme de transport B) saturation à fortes concentrations de substances d) la possibilité de transport contre le gradient de concentration d) une énergie considérable.
7. Concentration de la pharmacocinétique postule centrale du médicament sanguin - le paramètre principal pour contrôler l'effet thérapeutique. Tâches résolues sur la base de la connaissance de ce postulat.
Publicité centrale (dogme) Pharmacocinétique: La concentration de LV dans le plasma sanguin détermine (déterminer quantitativement) l'effet pharmacologique.
Dans la plupart des cas, le taux d'absorption, la distribution, le métabolisme et l'excrétion des médicaments sont proportionnels à leur concentration plasmatique sanguine (obéissent la loi des masses actuelles), donc il est possible:
1) Déterminer la période de semi-élimination (pour LS avec cinétique de premier ordre)
2) Expliquez la durée des effets toxiques de certains ls (pour les médicaments à fortes doses avec une cinétique de saturation)
[C] plasma → [c] sur la cible → effet
Défini par les lois sont déterminés par la pharmacie
Distributions
Tâches: Apprenez à contrôler la concentration de médicaments sanguins.
Modèles pharmacocinétiques (chambre à chambre unique et à deux chambres), lois quantitatives d'aspiration et d'élimination des médicaments.
L'organisme entier est un seul conteneur homogène. Hypothèses: 1) Le développement dynamique rapide est établi entre le contenu du médicament dans ... 2) LS rapidement et uniformément réparties dans tout le volume sanguin.Distribution de médicaments dans le corps. Compartiments, ligands. Les principaux déterminants de la distribution.
Compartiments de distribution: 1. Espace extracellulaire (plasma, fluide intercellulaire) 2. Cellules (cytoplasme, membrane organienne)Constante d'élimination, son essence, dimension, connexion avec d'autres paramètres pharmacocinétiques.
Taux d'élimination constante (K el, min -1) - montre quelle partie du médicament est éliminée du corps par unité de temps þ Kel \u003d un extérieur / un total, où et la quantité de LS, surlignée dans des unités. temps et le nombre total de LS dans le corps.
La valeur de K el est généralement constatée en résolvant une équation pharmacocinétique décrivant le processus d'élimination des médicaments du sang, donc k el s'appelle l'indicateur de modèle de la cinétique. La relation directe avec la planification du mode posologique K el n'a pas, mais sa valeur est utilisée pour calculer d'autres paramètres pharmacocinétiques.
La constante d'élimination est directement proportionnelle au jeu et proportionnelle inversement au volume de la distribution (de la définition de la clairance): Kel \u003d CL / VD; \u003d heure -1 / min -1 \u003d part par heure.
La demi-vie des drogues, son essence, sa dimension, ses relations avec d'autres paramètres pharmacocinétiques.
Période de semi-élimination (T ½, min) est le temps nécessaire pour réduire la concentration de LS dans le sang en douceur de la moitié. Dans le même temps, il ne joue pas le rôle de la manière dont la concentration est atteinte - avec l'aide de la biotransformation, de l'excrétion ou de la combinaison des deux processus.
La période de semi-élimination est déterminée par la formule:
![]()
Demi-vie - le paramètre pharmacocinétique le plus important qui permet:
b) déterminer le temps d'élimination complète du médicament
c) prédire la concentration de médicaments à tout moment (pour LS avec cinétique de premier ordre)
Dégagement en tant que paramètre principal de la pharmacocinétique pour contrôler le mode de dosage. Son essence, sa dimension et une connexion avec d'autres indicateurs pharmacocinétiques.
Autorisation (Cl, ml / min) - le volume de sang, qui est effacé des médicaments par unité de temps.
Parce que Plasma (sang) - une partie "visible" du volume de distribution, puis de dédouanement - fraction du volume de distribution, à partir duquel le médicament est distingué par unité de temps. Si vous désignez la quantité totale de médicaments dans le corps à travers Et communet le montant qui a été médié par Et la sortie, ensuite:
D'autre part, de déterminer la quantité de distribution, il s'ensuit que la quantité totale de médicaments dans le corps est Et général \u003d V dés / plasma. En substituant cette valeur dans la formule de dédouanement, nous obtiendrons:
![]() .
.
Ainsi, le dégagement est le rapport de la vitesse de la drogue à sa concentration dans le plasma sanguin.
Dans ce formulaire, la formule de dédouanement est utilisée pour calculer la dose de maintenance du médicament ( D p.), C'est-à-dire la dose du médicament qui devrait compenser la perte de médicaments et soutenir son niveau à un niveau constant:
Introduction Vitesse \u003d Vitesse électorale \u003d CL'C TER (dose / min)
D n \u003d vitesse d'utilisation (T - intervalle, entre la consommation de drogue)
Additif de dégagement. L'élimination de la substance du corps peut survenir avec la participation des processus dans les reins, les poumons, le foie et d'autres organes: CL systémique \u003d cl Renis. + CL LEVER + CL DR.
La clairance est connectée avec une période de semi-élimination des médicaments et de la distribution: T 1/2 \u003d 0,7 * vd / cl.
Dose. Types de doses. Unités de dosage de médicaments. Objectifs de dosage, méthodes et options d'introduction, intervalle d'introduction.
L'effet des médicaments sur le corps est largement déterminé par leur dose.
Dose - la quantité de substance introduite dans l'organisme dans une réception; Il est exprimé en unités de poids, en vrac ou conditionnel (biologique).
Types de doses:
a) dose unique - la quantité de substance pour une réception
b) dose quotidienne - la quantité de médicament assignée à une ou plusieurs réceptions par jour
c) Dose à terme - la quantité totale du médicament pour le traitement
d) doses thérapeutiques - doses dans lesquelles le médicament est utilisé avec thérapeutique ou cibles préventives (seuils ou doses thérapeutiques minimales, moyenne thérapeutique et supérieure).
e) Doses toxiques et mortelles - Doses de LV, dans lesquelles ils commencent à avoir des effets toxiques prononcés ou à causer la mort du corps.
e) la dose de chargement (entrée) - le nombre de LS injecté, qui remplit tout le volume de la distribution du corps dans la concentration active (thérapeutique): DR \u003d (CSS * VD) / F
g) Dose de soutien - Saisie systématique du nombre de médicaments compensant les pertes LS avec dégagement: PD \u003d (CSS * cl * dt) / F
Unités de dosage Pls:
1) dans les grammes ou les fractions de grammes
2) Nombre de médicaments par 1 kg masses corporelles (par exemple, 1 mg / kg) ou par unité de surface corporelle (par exemple, 1 mg / m 2)
Buts de dosage LS:
1) Déterminer le nombre de médicaments nécessaires pour causer la volonté souhaitée effet thérapeutique Avec une certaine durée
2) Évitez les phénomènes d'intoxication et d'effets secondaires avec l'introduction de médicaments
Méthodes pour l'introduction de HP: 1) Enteral 2) Parentéral (voir. 5)
Variantes d'administration de la LS.:
a) Continu (par perfusion intravasculaire prolongée ls goutte à goutte ou via des distributeurs automatiques). Avec une introduction continue de médicaments, sa concentration dans le corps varie en douceur et n'est pas soumise à des fluctuations importantes
b) Administration intermittente (méthodes d'injection ou d'injection) - l'introduction de médicaments à certains intervalles (intervalles de dosage). Avec une introduction discontinue de médicaments, sa concentration dans le corps fluctue continuellement. Après avoir reçu une certaine dose, il augmente initialement, puis diminue progressivement, atteignant des valeurs minimales avant la prochaine administration du médicament. Les oscillations de la concentration sont les plus importantes que la plus grande que la dose de médicament et l'intervalle entre les introductions.
Intervalle d'introduction - l'intervalle entre les doses administrées, assurant le maintien de la concentration thérapeutique de la substance dans le sang.
15. L'introduction de médicaments à une vitesse constante. La cinétique de la concentration du médicament dans le sang. Concentration stationnaire du médicament dans le sang (C SS), le temps de sa réalisation, de son calcul et de sa gestion.
 La particularité de l'introduction de HP avec une vitesse constante est un changement en douceur de sa concentration de sang lorsqu'il est administré, tandis que:
La particularité de l'introduction de HP avec une vitesse constante est un changement en douceur de sa concentration de sang lorsqu'il est administré, tandis que:
1) Le temps nécessaire pour atteindre la concentration de médicaments stationnaires est de 4-5t ½ et ne dépend pas du taux d'infusion (la valeur de la dose administrée)
2) avec une augmentation du taux d'infusion (dose administrée), la valeur avec SS augmente également d'un nombre proportionnel
3) Éliminer les médicaments du corps après la fin de la perfusion occupe 4-5t ½.
CSS - Concentration stationnaire Equilibrium - la concentration de médicaments atteints au taux d'administration de la vitesse égale de retrait, de sorte:
![]() (de la définition du dédouanement)
(de la définition du dédouanement)
Pour chaque demi-vie ultérieure, la concentration de LS augmente sur la moitié de la concentration restante. Tous ls, obéissant à la loi de l'élimination de la première commande, atteindra CSS après 4-5 demi-vie.
Approches de gestion de niveau SSS: Changez la dose de LS ou de l'intervalle d'administration
16. Administration des médicaments intermittents. Cinétique de la concentration de la drogue dans le sang, la gamme thérapeutique et toxique de concentrations. Le calcul de la concentration fixe (C SS), les limites de ses oscillations et le contrôle. Un intervalle adéquat de doses discrètes.
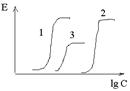
 Fluctuations de la concentration de LS dans le plasma sanguin: 1 - avec une administration gouttière intraveineuse constante; 2 - par une administration fractionnée de la même dose quotidienne à un intervalle de 8 heures; 3 - avec l'introduction d'une dose quotidienne avec un intervalle de 24 heures.
Fluctuations de la concentration de LS dans le plasma sanguin: 1 - avec une administration gouttière intraveineuse constante; 2 - par une administration fractionnée de la même dose quotidienne à un intervalle de 8 heures; 3 - avec l'introduction d'une dose quotidienne avec un intervalle de 24 heures.
Introduction intermittente LS. - Introduction certain nombre Ls à intervalles.
La concentration stationnaire à l'équilibre est réalisée grâce à 4 à 5 périodes de semi-élimination, le temps de sa réalisation ne dépend pas de la dose (au début, lorsque le niveau de concentration de LS est faible, le taux de son élimination est également faible; Comme le montant de sa substance augmente, le taux de son élimination augmente, tellement tôt ou cela arrivera en retard un tel moment où l'augmentation du taux d'élimination équilibre la dose administrée de médicaments et l'augmentation supplémentaire de la concentration pour arrêter)
Le CSS est directement proportionnel à la dose de médicaments et proportionnelle inversement à l'intervalle d'introduction et de dédouanement des médicaments.
Borders oscillation CSS: ![]() ; C ss min \u003d c ss max × (1 - el. Fr.). Les fluctuations de la concentration de la LS sont proportionnelles à T / T 1/2.
; C ss min \u003d c ss max × (1 - el. Fr.). Les fluctuations de la concentration de la LS sont proportionnelles à T / T 1/2.
Gamme thérapeutique (couloir de sécurité, fenêtre thérapeutique) - Il s'agit d'un intervalle de concentrations de la thérapeutique minimale à l'apparition des premiers signes d'effets secondaires.
Gamme toxique - L'intervalle de concentration du plus haut thérapeutique à la mort.
Mode adéquat d'introduction de doses discrètes: Ce mode d'administration dans lequel la fluctuation de la concentration du médicament dans le sang est placée dans la plage thérapeutique. Pour déterminer le mode d'administration adéquat du médicament, il est nécessaire de calculer. La différence entre CSS max et CSS min ne doit pas dépasser 2CSS.
Gestion des oscillations CSS:
Les échappements des oscillations CSS sont directement proportionnelles à la dose de médicaments et sont inversement proportionnelles à l'intervalle de son introduction.
1. Changer la dose de HP: Avec une augmentation de la dose de drogues, la plage d'oscillation que son CSS augmente proportionnellement
2. Changer l'intervalle d'imposition du réseau local: Avec une augmentation de l'intervalle d'administration de la LS, la plage d'oscillation de son CSS est proportionnelle à la diminution
Dans le même temps, changez la dose et l'intervalle de l'introduction
Dose d'introduction (chargement). Signification thérapeutique, calcul sur les paramètres pharmacocinétiques, les conditions et les restrictions sur son utilisation.
Dose d'introduction (chargement) - Dose administrée en une seule réception et remplissant l'ensemble du volume de la distribution dans la concentration thérapeutique active. VD \u003d (CSS * VD) / F; \u003d mg / l, \u003d l / kg
Signification thérapeutique: La dose d'introduction fournit rapidement la concentration thérapeutique agissante de médicaments dans le sang, ce qui permet, par exemple, d'arrêter rapidement l'attaque, l'arythmie, etc.
La dose d'introduction peut être introduite à la fois uniquement lorsque ignoré le processus de distribution de la substance
Restreindre l'utilisation du mercredi: Si la distribution de LS se produit beaucoup plus lentement que son flux sanguinL'introduction de l'ensemble de la dose de démarrage à la fois (surtout par voie intraveineuse) créera une concentration de manière significative au-dessus de la thérapeutique et provoque la survenue d'effets toxiques. La condition d'utiliser le WD: Par conséquent, l'introduction de doses de charge devrait toujours être lent ou fraction.
Soutenir des doses, leur signification thérapeutique et leur calcul pour le mode de dosage optimal.
Signification thérapeutique: PD compense la perte de la clienne pour l'intervalle entre les introductions du médicament. Calcul pour une distribution optimale des médicaments (pour une liaison rapide de l'attaque): ... 1. Calculez le VD: DR \u003d (CSS * VD) / FIndividuel, âge et différences sexuelles de la pharmacocinétique des médicaments. Amendements visant à calculer les valeurs individuelles du volume de distribution des médicaments.
2. Différences sexuelles dans l'action des médicaments. Pour les femmes, un poids corporel plus petit est caractérisé que chez les hommes, donc la valeur de la dose du médicament pour eux devrait ... 3. Conditions pathologiques L'organisme et l'effet des médicaments a) Maladie du foie: F médicaments en raison de la fermeture du métabolisme pratiqué, une fraction de LS sans bornes en raison du manque de ...Liquidation des drogues, mécanismes, leurs caractéristiques quantitatives et qualitatives.
Mécanismes de dédouanement rénal et de leur caractéristique: 1. Filtrage: LS, la surbrillance avec filtrage (insuline) aura une autorisation, ... est déterminée par: flux sanguin rénal, fraction LC non liée et capacité de filtration du rein.Facteurs affectant la clairance rénale des médicaments. La dépendance de la clairance des propriétés physicochimiques des médicaments.
a) filtrage glomérulaire B) débit sanguin rénal B) vitesse maximale de sécrétionDégagement hépatique des médicaments, ses déterminants et restrictions. Cycle de drogue entérogènes.
1) Métabolisme (biotransformation) par oxydation, restauration, alkylation, hydrolyse, conjugaison, etc. Stratégie principale du métabolisme de xénobiotiques: substances non polaires ® polaire ... 2) sécrétion (retrait des substances non traditionnelles en bile)Correction du traitement médicamenteux dans les dommages causés au foie et à d'autres conditions pathologiques.
Correction du régime de dosage de la maladie rénale, voir ci-dessus en B.26, principes généraux Correction - B.25. Correction du régime de dosage sous le contrôle de la clairance générale du médicament: correction de la dose: dind. \u003d DuttyN. × Clant. / Clutpichn.Correction du mode de dosage sous le contrôle de la fonction rénale résiduelle.
Nous savons: a) la fonction rénale résiduelle définissant la clairance de la créatinine dans cette ... B) Clairance générale de ce médicament (CLA / Total) et la part de la clairance rénale des médicaments en dégagement généralStratégie de traitement médicinale individuelle.
Confession un rôle important Concentration comme lien de liant La pharmacocinétique et la pharmacodynamique contribuent à la création d'une stratégie de concentration cible - optimisation de la dose dans un patient donné basé sur la mesure de la concentration de LS. Il se compose des étapes suivantes:
1. Sélection de la concentration de la cible
2. Calcul de V D et CL basé sur des valeurs typiques et des amendements afin de prendre en compte de tels facteurs que le poids corporel et la fonction rénale.
3. Entrer dans la dose de démarrage ou la dose de maintenance, calculée sur la base des valeurs de TC, V D et CL.
4. Enregistrement de la réaction du patient et la définition de la concentration de LS
5. Examiner V D et CL en fonction des résultats de la mesure de la concentration.
6. Répétez les étapes 3-6 dans le but de choisir la dose nécessaire requise pour la réaction optimale.
La biotransformation des médicaments, son sens biologique, l'orientation principale et l'effet sur l'activité des médicaments. Les principales phases des transformations métaboliques de médicaments dans le corps.
Biotransformation de drogues - transformation chimique des médicaments dans le corps.
Signification biologique de la biotransformation des médicaments: créer un substrat pratique pour une élimination ultérieure (en tant que matériau énergétique ou plastique) ou dans l'accélération de l'élimination de la LS du corps.
Les transformations métaboliques de médicaments: métabolites polaires → polaires (hydrophiles) affichées avec l'urine.
Les deux phases des réactions métaboliques des médicaments sont distinguées:
1) Transformation métabolique (réactions instinctives, phase 1) - Conversion des substances dues à l'oxydation microsomale et éminomique, la récupération et l'hydrolyse
2) conjugaison (réactions synthétiques, phase 2) - processus biosynthétique, accompagné d'un ajout au médicament ou de ses métabolites d'un certain nombre de groupes chimiques ou de molécules de composés endogènes par a) la formation de glucuronides b) des esters de glycérol) sulfoockets d ) acétylation d) méthylation
L'influence de la biotransformation sur l'activité pharmacologique de la LAN:
1) Le plus souvent, les métabolites de la biotransformation ne possèdent pas une activité pharmacologique ou leur activité est réduite par rapport à la matière de départ
2) Dans certains cas, les métabolites peuvent maintenir une activité et même dépasser l'activité de la substance source (la codéine est métabolisée à une morphine plus active pharmacologique)
3) Parfois, des substances toxiques sont formées pendant la biotransformation (métabolites d'isoiazide, lidocaïne)
4) Parfois, des métabolites avec des propriétés pharmacologiques opposées sont formées lors de la biotransformation (métabolites d'agonistes non sélectifs B 2 - adrénorécepteurs possèdent les propriétés de ces bloqueurs de récepteurs)
5) Un certain nombre de substances sont des prodrugs qui ne donnent initialement pas d'effets pharmacologiques, mais pendant la biotransformation est convertie au BAV (inactif L-Dope, pénétré par la BC, se transforme en un cerveau en dopamine active, tandis qu'il n'y a pas d'effets systémiques de dopamine).
La signification clinique de la biotransformation des médicaments. Facteurs affectant leur bootransformation. Interaction métabolique des médicaments.
Influence sur la biotransformation de ls divers facteurs: a) état fonctionnel du foie: avec ses maladies du dédouanement LS habituellement ... B) L'effet des facteurs environnementaux: le tabagisme contribue à l'induction du cytochrome P450, à la suite de laquelle le métabolisme des drogues ...Façons et mécanismes d'élimination des médicaments du corps. Possibilités de gérer la drogue.
Chemins et mécanismes d'élimination des médicaments:Élimination du foie et des reins de la LAN et d'autres organes:
a) reins par filtration, sécrétion, réabsorption
b) foie par biotransformation, excrétion avec bile
c) à travers les poumons, la salive, la sueur, le lait, etc. par sécrétion, évaporation
Les possibilités de gérer les processus de suppression des médicaments:
1. Contrôle du pH: Dans l'urine alcaline, l'élimination des composés acides augmente, en élimination acide des composés principaux
2. Application drogues cholérétiques (Holenzim, Allohol)
3. Hémodialyse, dialyse péritonéale, hémosorption, lymphosorption
4. Dirésis forcés (v / dans NaCl ou glucose pour charge d'eau + Furosémide ou mannitol)
5. Laver l'estomac, appliquer un lavement
Le concept de récepteurs en pharmacologie, la nature moléculaire des récepteurs, les alarmes de l'action des médicaments (types d'alarme transmembranaire et intermédiaires secondaires).
Récepteurs -composants moléculaires d'une cellule ou d'un organisme qui interagissent avec des médicaments et induisent un certain nombre d'événements biochimiques menant au développement de l'effet pharmacologique.
Le concept de récepteurs en pharmacologie:
1. Les récepteurs déterminent les modèles quantitatifs de l'action LS
2. Les récepteurs sont responsables de la sélectivité de l'action LS
3. Intermédiaires des récepteurs d'antagonistes pharmacologiques
Le concept de récepteurs - la base de l'utilisation initiale des médicaments affectant les processus et les communications réglementaires, biochimiques.
Nature du récepteur moléculaire:
1. Protéines réglementaires, médiateurs d'action de divers signaux chimiques: neurotransmetteurs, hormones, autocoïdes
2. Porte-protéines enzymes et protéines transmembranaires (Na +, K + ATPAZ)
3. Protéines structurelles (tubuline, protéines cytosquelette, surface cellulaire)
4. Protéines nucléaires et acides nucléiques
Mécanismes de signalisation d'action antidrogue:
1) Pénétration de ligand soluble dans les lipides à travers la membrane et leur effet sur les récepteurs intracellulaires.
2) La molécule de signal se lie au domaine extracellulaire de la protéine transmembranaire et active l'activité enzymatique de son domaine cytoplasmique.
3) La molécule de signal est associée au canal d'ions et ajuste son ouverture.
4) La molécule de signal se lie au récepteur de la surface de la cellule, qui est conjugué avec l'enzyme effecteur par la protéine G. G-protéine active le médiateur secondaire.
Types d'alarme transmembranaire:
a) à travers des récepteurs de 1 TMS possédant et ne possédant pas une activité de tyrosine kinase
b) à travers des récepteurs 7-TMS associés à la protéine G
c) à travers des canaux d'ions (contacts fendus à charge potentiels, dépendants du ligand)
Intermédiaires secondaires: Tsamf, ions CA2 +, Dag, if3.
Mécanismes physico-chimiques et chimiques de l'action des substances médicinales.
Les principaux effets pharmacologiques: 1) Narcotic 2) Dépressives générale 3) Paralysing 4) Action de membronolytique 5). Substances naturelles chimiques: hydrocarbures inertes chimiquement, éthers, alcools, ... Mécanisme d'action - membranes de dzing réversibles.Sélectivité et spécificité de l'action des médicaments. Les effets thérapeutiques, latéraux et toxiques de la drogue, leur nature du point de vue du concept de récepteurs. Stratégie thérapeutique de lutte contre les effets secondaires et toxiques de la drogue.
Spécificité - Bossing LS avec un type de récepteur strictement spécifique.
Sélectivité - Il est capable de lier un réseau local avec un ou plusieurs types de récepteurs plus précisément qu'aux autres.
Plus de préférence utiliser la sélectivité du terme, car Il est peu probable que toute molécule LS ne puisse être mise en contact avec un type de molécules de récepteur, car le nombre de récepteurs potentiels chez chaque patient a une importance astronomique.
Action thérapeutique - L'effet pharmacologique principal souhaité, attendu de ce médicament pharmacologique.
Effets secondaires- les effets qui se produisent avec l'utilisation de substances de doses thérapeutiques et constituent le spectre de leur action pharmacologique.
Effets toxiques - des effets indésirables qui se manifestent dans cette HP lorsqu'ils quittent la gamme thérapeutique.
Communication d'effets thérapeutiques et toxiques des médicaments sur la base de l'analyse des mécanismes de récepteur et d'effecteur:
1) les effets thérapeutiques et toxiques médiés par le même mécanisme de récepteur-effecteur (la prazosine agit comme un antagoniste alpha-sélectif pour les récepteurs des navires GMC et a un effet hypotenseur dans l'hypertension essentielle, mais au cours de sa grande dose, un patient peut avoir une hypotension posturale)
2) Effets thérapeutiques et toxiques médiés par des récepteurs identiques, mais différents tissus ou divers chemins effecteurs (glycosides cardiaques sont utilisés pour augmenter la capacité contractile du myocarde, en même temps, ils violent la fonction du tractus gastro-intestinal, en raison du blocus de la membrane cellulaire NA + / K +)
3) Effets thérapeutiques et toxiques médiés divers types Les récepteurs (par exemple, la norépinéphrine a un effet hypertenseur à travers un 1 -ar, mais il provoque la tachycardie à travers B 1 -ar)
Stratégie thérapeutique de lutte contre la thérapeutique et effets secondaires Ls:
1. LAN doit toujours être administré dans la dose la plus basse qui provoque un effet thérapeutique acceptable.
2. réduire la dose d'un médicament en raison de la nomination d'un autre réseau local avec effet similaireMais à travers d'autres récepteurs et avec un profil de toxicité différent.
3. La sélectivité de l'action LS peut être augmentée en contrôlant la concentration de LS dans la zone du récepteur de différentes parties du corps ( application locale LS - Utilisation d'inhalation du salbutamol avec asthme bronchique)
32. Termes et concepts de pharmacologie quantitative: effet, efficacité, activité, agoniste (complet, partiel), antagoniste. Différence clinique des concepts Activité et efficacité des médicaments.
 Effet (réponse)- le rendement quantitatif de la réaction de l'interaction de la cellule, de l'organe, du système ou du corps avec un agent pharmacologique.
Effet (réponse)- le rendement quantitatif de la réaction de l'interaction de la cellule, de l'organe, du système ou du corps avec un agent pharmacologique.
Efficacité - mesure de la réaction le long de l'axe d'effet - la valeur de la réponse du système biologique à l'effet pharmacologique; c'est la capacité des médicaments de rendre l'action maximale possible pour cela.. Ceux. En fait, il s'agit de l'effet maximal de l'effet pouvant être obtenu lorsque ce médicament est introduit. Caractérisé numériquement par la valeur de e max. Plus le max est élevé, plus l'efficacité du médicament est élevée.
Activité - La mesure de la sensibilité aux médicaments le long de l'axe de concentration caractérise l'affinité (affinité du ligand au récepteur) montre comment la dose (la concentration) du médicament est capable de développer un effet standard égal à 50% du médicament maximal possible. Normalement caractérisé par l'UE 50 ou ED 50. Plus l'activité de médicaments est élevée, plus la dose est réduite pour reproduire l'effet thérapeutique.
Efficacité: 1 \u003d 2\u003e 3
Activité: 1\u003e 3\u003e 2
Dans les activités cliniques, il est plus important de connaître l'efficacité et non l'activité, car Nous sommes plus intéressés par la capacité du réseau local à causer une action spécifique dans le corps.
Agoniste- Ligand, qui lie au récepteur et provoque une réponse biologique, déclenchant le système physiologique. Agoniste complet- réponse maximale, partiel - provoquer une réaction plus petite même lorsque tous les récepteurs occupent.
 Antagoniste - Les ligands occupent des récepteurs ou les modifient de manière à perdre la capacité d'interagir avec d'autres ligands, mais ne provoquent pas de réaction biologique (action block d'agonistes).
Antagoniste - Les ligands occupent des récepteurs ou les modifient de manière à perdre la capacité d'interagir avec d'autres ligands, mais ne provoquent pas de réaction biologique (action block d'agonistes).
Antagonistes compétitifs- Interagissez avec les récepteurs de manière réversible et concurrentiellement des agonistes. Une augmentation de la concentration d'agoniste peut complètement éliminer l'effet antagoniste. L'antagoniste compétitif déplace la courbe "dose-effet" pour l'agoniste, augmente 50 CE 50, n'affecte pas e max.
Antagonistes non concurrentiels- modifier de manière irréversible l'affinité des récepteurs à l'agoniste, la liaison ne se produit souvent pas avec le secteur actif du récepteur, une augmentation de la concentration de l'agoniste n'élimine pas l'action de l'antagoniste. L'antagoniste non concurrentiel réduit EMAX, ne change pas le CE50, la courbe "dose-effet" est compressée par rapport à l'axe vertical.
33. Lois quantitatives de l'action des médicaments. Loi de réduire la réponse des systèmes biologiques. Modèle Clark et sa conséquence. Forme générale La dépendance à la concentration est l'effet dans les coordonnées normales et lognormales.
Clark-Ariens Modèle:
1. L'interaction entre le ligand (L) et le récepteur (R) est réversible.
2. Tous les récepteurs de ce ligand sont équivalents et indépendants (leur saturation n'affecte pas les autres récepteurs).
3. L'effet est directement proportionnel au nombre de récepteurs occupés.
4. Le ligand existe dans deux états: libre et associé au récepteur.
A), où KD est une constante d'équilibre, une activité interne KE.
B) parce que En cas d'augmentation du nombre de ligands à un moment donné, tous les récepteurs seront occupés, le nombre maximal possible de complexes récepteurs de ligand éduqués est décrit par la formule:
\u003d [R] × (1)
L'effet est déterminé par la probabilité d'activation du récepteur lors de la liaison au ligand, c'est-à-dire Son activité interne (KE), donc e \u003d ke ×. Dans ce cas, l'effet est maximum à clé \u003d 1 et minimal et ke \u003d 0. Naturellement, que effet maximum Décrit par le rapport EMAX \u003d KE ×, où - le nombre total de récepteurs pour ce ligand
L'effet dépend de la concentration de ligand sur les récepteurs [S], donc
Des ratios réduits, il s'ensuit que CE 50 \u003d KD
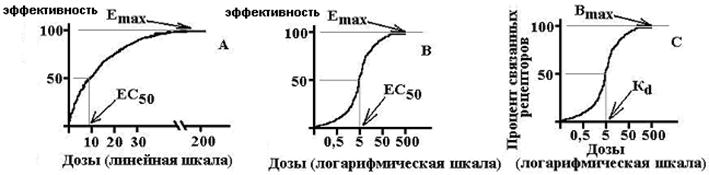
E max est l'effet maximum, B Max est le nombre maximal de récepteurs connexes, EC 50 est la concentration de la LS, dans laquelle l'effet se produit égal à la moitié du maximum, K D est la constante de dissociation du récepteur à laquelle 50% de Les récepteurs sont associés.
La loi de la diminution de la réponseconforme à la dépendance parabolique «Concentration - efficacité». La réponse aux petites doses de drogue augmente généralement directement proportionnelle à la dose. Toutefois, avec une augmentation de la dose, la croissance de la réponse est réduite et finalement une dose peut être obtenue, à laquelle il n'ya plus d'augmentation de la réponse (en raison de l'occupation de tous les récepteurs de ce ligand).
Effet d'effets gradués et quantiques, Essence et applications cliniques. Évaluation quantitative de l'activité et de l'efficacité des médicaments dans la pratique expérimentale et clinique.
Tous les effets pharmacologiques peuvent être divisés en deux catégories:
mais) effets graduels (continus, intégrales) - de tels effets des médicaments pouvant être mesurés quantitativement (l'effet des médicaments anti-hypotenseurs - au niveau de la pression artérielle). Décrit la "courbe de dose-effet" progressive (voir 33), sur la base desquelles il est possible d'estimer: 1) la sensibilité individuelle à la drogue; 2) activité ls; 3) efficacité maximale des médicaments.
b) effets quantiques - de tels effets des médicaments, qui sont une valeur discrète, une caractéristique qualitative, c'est-à-dire Il est décrit en quelques variations d'états (mal de tête après avoir reçu des analgésiques ou non). La courbe quantique de la dose-effet est décrite, où la dépendance de l'effet dans la population de la valeur de la dose reçue de LS est décrite. Le graphique de la dépendance à effet de dose a une forme en forme de dôme et est identique à la courbe gaussienne de la distribution normale. Sur la base de la courbe quantique, il est possible: 1) Évaluer la sensibilité de la population des médicaments; 2) Notez l'effet de l'effet à une dose donnée; 3) Choisissez une dose thérapeutique moyenne.
Différences entre la caractéristique progressive et quantique de "dose-effet":
L'évaluation quantitative de l'activité et de l'efficacité de LS est effectuée sur la base de la construction des courbes «dose-effet» et de leur évaluation ultérieure (voir B.32)
Types d'action de la drogue. Changer l'action des médicaments pendant leur réutilisation.
1. Localisation: · Local - Action qui se produit sur le site de l'application PLA (généralement en cuir et ... · Resorbant est une action que la LS doit être absorbée dans la circulation sanguine ou directement introduite dans ...La dépendance de l'action des médicaments d'âge, de genre et de capacités individuelles du corps. La valeur des rythmes quotidiens.
· Enfants - la zone de pharmacologie, engagée dans l'étude des caractéristiques de l'action des médicaments sur organisme pour enfants, Appelée pharmacologie pédiatrique. ... · personnes âgées - chez les personnes âgées et âgé Pharmacocinétique ... 2. Paul - Des expériences animales et des observations cliniques indiquent qu'il existe des différences sexuelles de ...Variabilité et variabilité de l'action de la drogue. Hypo- et hyperréactivité, tolérance et tachofilaxie, hypersensibilité et idiosyncrasie. Causes de la variabilité des médicaments et de la stratégie de thérapie rationnelle.
La variabilité de l'action LS - reflète la différence entre les effets pharmacologiques d'une drogue parmi les différentes personnes.
La variabilité de l'action des médicaments - reflète la capacité du réseau local à fournir une action différente de ses effets pharmacologiques typiques.
Variantes de variabilité et de variabilité de l'action LAN:
1. Hyporactivité - réduire l'effet de cette dose de médicaments par rapport à l'effet observé chez la plupart des patients.
2. Hyperréactivité - Améliorer l'effet de cette dose de médicaments par rapport à l'effet observé chez la plupart des patients.
3. Tolérance - réduction de la réponse de l'organisme sur l'introduction répétée de médicaments; Afin de restaurer la réaction au réseau local, il est nécessaire d'introduire de plus en plus de doses.
4. Tahofilaxie - une condition dans laquelle l'introduction fréquente de la LS provoque le développement de la tolérance après quelques heures, mais à une injection suffisamment rare de médicaments, son effet est entièrement maintenu. Le développement de la tachocilaxie est généralement associé à l'épuisement des systèmes effectrices.
5. Hypersensibilité - développement d'une réponse immunitaire allergique ou autre à la réinitialisation du réseau local.
6. Idiosyncrasie - la réponse pervers de l'organisme sur la LS associée aux caractéristiques génétiques des métabolismes de médicaments ou de réactivité immunologique individuelle.
Les principales raisons de la variabilité de l'action des médicaments:
1) Changement de la concentration de la substance dans la zone de récepteur - en raison des différences de taux d'aspiration, de sa distribution, de son métabolisme, de l'élimination
2) Variations de la concentration de ligand endogène - le récepteur-propranolol (β-adrénoblocator) ralentit la fréquence cardiaque chez les personnes avec niveau augmenté Les catécholamines dans le sang, mais n'affectent pas la fréquence cardiaque de fond des athlètes.
3) Modifiez la fonction de densité ou de récepteur.
4) Modification des composants de la réaction située distale que le récepteur.
La stratégie rationnelle de thérapie: c'est que, dans le but de la drogue, de sorte qu'il ait l'effet attendu, il est nécessaire de prendre en compte des facteurs individuels du corps (âge, sexe, etc.), les rythmes quotidiens de la personne, la présence de maladies chroniques et d'autres écarts. Il est également nécessaire de prédire le développement d'effets indéterminés, compte tenu de tout options possibles la variabilité du réseau local d'action.
Évaluation de la sécurité de la médecine. Index thérapeutique et limites de sécurité standard.
D'énormes outils sont dépensés pour la création et l'introduction d'un nouveau médicament - de 100 $ à 350 millions de dollars et plus encore. Ces dépenses comprennent le travail dépensé sur ... Afin de transmettre toutes les étapes de l'évaluation des médicaments, elle doit correspondre à la principale ... La sécurité des nouveaux LS est effectuée en 2 étapes:Interactions pharmacocinétiques des médicaments (exemples).
L'interaction pharmacocinétique LS est une variété d'interactions pharmacologiques, c'est-à-dire L'interaction des médicaments qui se manifeste uniquement avec leur admission articulaire au corps humain.
L'interaction pharmacocinétique est effectuée au stade de l'aspiration, de la distribution et du déposant, du métabolisme et de l'élimination.
1. À la phase d'aspiration - Ce type d'interaction peut entraîner une augmentation ou une diminution de leur aspiration. Il peut être évité si l'intervalle entre les récepteurs PLS est au moins 4 heures.
Avec l'administration du système d'exploitation, son absorption est déterminée:
· environnement de pH - Les médicaments non ionisés sont absorbés par le tractus gastro-intestinal mieux que ionisé que l'augmentation du pH du jus gastrique augmente l'absorption de bases faibles et réduit l'absorption des acides faibles. Exemple: Les antiacides, les blocateurs H 2-gismistamine ont poussé l'absorption du cétoconazole et d'autres médicaments antifongiques, les anticoagulants indirects, l'acide acétylsalicylique, les barbituriques (empêchent presque complètement leur action hypnotique); L'augmentation du pH du milieu améliore l'absorption du gibutide, accélère la dissolution de la coque de substances solubles intestinales.
· interaction directe dans le gastro-intestinal - La formation de complexes de chélate et de connexions ne suce pas dans le tractus gastro-intestinal. P ramers:les formes de carbone activées forment des composés insolubles avec des médicaments, empêchant leur aspiration dans l'intoxication; Les tétracyclines interagissent avec du calcium, de l'aluminium, du fer, du magnésium pour former des complexes de chélate, leur absorption diminue avec l'utilisation d'antiacides, préparations de bismuth; Fluoroquinolones + antiacides ou sucralfat \u003d diminution de l'efficacité de la thérapie antibactérienne.
· motoric Zhkt. - Peut causer une accélération ou ralentir l'aspiration de drogues. Exemples: Les procréateurs (méthoclopromide) accélèrent l'absorption de LS (éthanol, paracétamol, tétracycline) et ralentissement de l'aspiration lentement (digoxine, cimétidine); Laxatif signifie réduire l'aspiration et la biodisponibilité des médicaments; Lors de la prise de médicaments anticholinergiques, les blocators H 2-gismine récepteurs (allongement du temps de passage du réseau local sur le tractus gastro-intestinal) augmente la biodisponibilité et l'absorption des glycosides cardiaques, des préparations de fer, qui peuvent conduire à la manifestation d'effets toxiques.
· microflore intestinale - Il faut une participation directe à l'absorption des médicaments. Toute dysbactérite est donc manifestée par une absorption de la LS altérée. Exemples: Digoxine + érythromycine \u003d augmentation de la concentration de la digoxine dans le sang et le développement d'effets indésirables; Converse orale + AB d'un large spectre \u003d réduction de l'effet de la connexion
· dommages causés par les intestins - ralentit l'aspiration de certains LS. Exemples: cytostatique (cycloofvamide) inhiber l'absorption de la digoxine; Violation de l'absorption du fer, de la cyanocobalamine, de l'acide folique.
Au stade de la distribution et de la dépôt
· Déplacement concurrentiel de l'albumine plasmatique sanguine - Si le médicament est associé à des protéines inférieures à 90%, le déplacement ne le conduira pas à ... · Déplacement avec des protéines dans les tissus: la quinidine déplace la digoxine + ... 3. À La phase de métabolisme - LS peut augmenter ou réduire l'activité systèmes d'enzymeParticipé au métabolisme du réseau local (...Au stade de l'enlèvement
· Modification de la sécrétion de canal - Comté + Digoxine \u003d augmentation de la concentration de la digoxine du sang et le développement d'effets toxiques (comté de ... · Les modifications de la réabsorption des canaux - la réabsorption ne sont soumises à ... 40.Farmacodynamique interaction des substances médicinales. Antagonisme, Synergisme, leurs types. Caractère du changement ...Effets secondaires et toxiques des substances médicinales. Effet teratogénique, embrypoxique et mutagène des médicaments. Médecine I. aspects sociaux Lutte contre la toxicomanie, la toxicomanie et l'alcoolisme. Le concept de toxicomique.
Effets secondaires - ces effets qui se produisent lorsque l'utilisation de substances de doses thérapeutiques et constituent le spectre de leur action pharmacologique, peut être primaire et secondaire:
a) Effets secondaires principaux - comme une conséquence directe de l'influence ce médicament Sur un certain substrat (hypospalivation lors de l'application de l'atropine pour éliminer le bradiarhymium)
b) Effets secondaires secondaires - Effets indésirables émergents indirectement (AB, supprimant une microflore normale, peut entraîner une superinfection)
Effets toxiques - des effets indésirables qui se manifestent dans cette HP lors de la sortie de la gamme thérapeutique (surdose LS)
La sélectivité de l'action LAN dépend de sa dose. Plus la dose de la drogue est élevée, moins il devient des élections.
Action tératogène- la capacité de la drogue lors de la nomination de sa femme enceinte de causer des anomalies anatomiques pour le développement du fœtus (Talidomide: Focomelia, Antoblatural LS: Multiples défauts)
Action embryotoxique - Effets indésirables qui ne sont pas associés à la violation de l'organogenèse au cours des trois premiers mois de la grossesse. Sur plus heure tardive manifeste action féthotoxique.
Effet mutagénique de HP - Dommages causés à la cellule germinale et à son appareil génétique de médicaments, qui se manifeste par un changement du génotype de progéniture (adrénaline, cytostatique).
Action cancérogène ls. - la capacité de certains ls à induire la carcinogenèse.
1) La toxicomanie - l'état de la psyché et / ou l'état physiqueCe qui est une conséquence de l'impact sur le corps du réseau local et se caractérise par des réactions comportementales spécifiques, difficiles à retirer la LS pour obtenir un effet mental spécial ou éviter l'inconfort en l'absence d'un LC dans le corps. La toxicomanie est caractérisée par:
mais) dépendance psychologique - Développement de détresse émotionnelle lors de l'arrêt de la réception des médicaments. Une personne se sent vide, immergée dans la dépression, sentant un sentiment de peur, d'anxiété, son comportement devient agressif. Tous ces symptômes psychopathologiques se posent contre le fond des réflexions sur la nécessité de se présenter un LS, qui a provoqué une dépendance. Le désir de recevoir des médicaments peut varier de désir simple Avant la soif passionnée de la réception de la drogue, qui absorbe tous les autres besoins et se transforme en sens de la vie humaine. On pense que la dépendance psychologique se développe lorsqu'une personne apparaît une conscience que le bien-être optimal peut atteindre exclusivement grâce à l'introduction de HP. La Fondation dépendance psychologique - La foi humaine en action (dans la littérature, des cas de développement de la dépendance psychologique sur placebo).
b) dépendance physique - violation normale état physiologique Le corps qui nécessite une présence constante de médicaments pour maintenir l'état d'équilibre physiologique. La cessation de la consommation de drogue provoque le développement d'un complexe de symptômes spécifique - un syndrome abstineent - un complexe de troubles mentaux et neurovégétatifs sous la forme d'une fonction de la fonction sur le côté, en face de çaCe qui est caractéristique de l'action (la morphine élimine la douleur, opprime le centre respiratoire, rétrécit les élèves, provoque la constipation; pendant le patient, le patient a une douleur douloureuse, une respiration bruyante fréquente, des élèves sont élargis et développant une diarrhée résistante)
dans) tolérance. La tolérance aux moyens causant une dépendance à la drogue est souvent transversale, c'est-à-dire Il se produit non seulement à ce composé chimique, mais également à tous les composés structurellement similaires. Par exemple, chez les patients atteints de dépendance médicinale à la morphine, la tolérance se produit non seulement, mais également à d'autres analgésiques opioïdes.
Pour le développement de la toxicomanie, la présence de tous les 3 critères n'est pas prérequis.
Les opioïdes, les barbituriques, l'alcool causent de graves dépendances physiques, psychologiques et de tolérance. Anxiolitiques (diazépam, alprazolam) causer de préférence une dépendance psychologique.
2) Dépendance à la drogue (dépendance narcotique)- Il s'agit d'une forme extrêmement sévère de toxicomanie, de l'utilisation compulsif des médicaments, caractérisée par une imposition croissante et irrésistible à l'introduction de ce médicament, augmentant ainsi sa dose. La compulsivité de l'attraction signifie que la nécessité de l'introduction de médicaments domine le patient sur tous les autres (même vitaux). De la position cette définition, Attraction à la morphine - La dépendance, tandis que l'attraction de la nicotine est une dépendance à la drogue.
3) La toxicomanie - caractérise une attraction moins intense pour la réception des médicaments, lorsque le rejet de la drogue ne provoque que sensation de lumière inconfort, sans le développement d'une dépendance physique ou d'une image détaillée de la dépendance psychologique. Donc La dépendance couvre cette partie de la dépendance à la drogue qui ne relève pas de la définition de la toxicomanie. Par exemple, la toxicomanie mentionnée ci-dessus est la nicotine - une forme de dépendance.
4) Abus de drogue - utilisation non autorisée de médicaments de ces doses et de telles manières qui diffèrent des normes médicales ou sociales acceptées dans cette culture et dans ce temps. Donc L'abus ne couvre que des aspects sociaux de la consommation de drogues. Un exemple d'abus est l'utilisation de stéroïdes anabolisants dans des sports ou d'améliorer le physique des jeunes hommes.
5) Alcoolisme - abus d'alcool chronique (alcool éthylique), menant de temps à des dommages à un certain nombre d'organes (foie, tractus gastro-intestinal, CNS, système cardiovasculaire, système immunitaire) et accompagné d'une dépendance psycho-physique.
6) Toxicomie - Abus chronique de divers LS (y compris des médicaments, de l'alcool, des hallucinogènes), manifesté par divers troubles mentaux et somatiques, violation du comportement, dégradation sociale.
Traitement médicinal Tâche dure et ingrat. Toujours pas créé technique efficacequi garantirait le succès du traitement de plus de 30 à 40% des patients. RÉALISATION N'importe quoi résultats notables Peut-être seulement avec une coopération complète des efforts du patient, un médecin et environnement socialdans lequel le malade (principe de volontaire et d'individualité). Basé sur méthodes modernes Lycée les principes suivants:
Méthodes psychothérapeutiques et d'emploi;
Traitement et réhabilitation du groupe (alcooliques anonymes, toxicomanes)
Abolition progressive ou forte du médicament contre le fond de la thérapie de désintoxication
Thérapie de remplacement (remplacement narcotique Analogues lents et durables avec leur annulation ultérieure; Par exemple, appelé. Le programme de thérapie de substitution de méthadone dans les toxicomanes d'héroïne)
Traitement avec des antagonistes spécifiques (naloxone et nantlexone) ou des moyens de sensibilisation (Teturam)
Méthodes neurochirurgicales de cryodécurcissement de la ceinture pain d'épice et d'hippocampe
42. INTERACTION PARAISÉTIQUE DES MÉDICAMENTS. Prévenir et précautions lors de la conduite de thérapie à perfusion.
Interaction pharmaceutique -le type d'interaction associé à une réaction physicochimique entre les médicaments dans le processus de préparation de médicaments avant l'introduction de ces fonds dans le corps humain
mais) erreurs typiquesconduisant à une incompatibilité pharmaceutique: la décharge de recettes complexes, un stockage incorrect ne tient pas compte de la possibilité d'adsorption de médicaments à la surface des plastiques (nitrates biologiques)
b) Problèmes liés à la perfusion: mélange de sels solubles, de dérivés d'acides faibles insolubles ou de bases entraînent leur précipitation; Les glycosides cardiaques et les alcaloïdes sont hydrolysés sous forme de dosage liquide; PH du milieu (alcaloïdes précipités de milieu alcalin)
c) Recommandations: 1) Tous les mélanges sont préférables de préparer EX Tempore 2) La solution la plus fiable avec un réseau local 3) Toutes les solutions doivent être vérifiées pour la présence de STRIPS 4) L'interaction peut se produire sans changement visible de solutions 5) n'est pas possible d'ajouter des médicaments au sang et aux solutions d'AK 6) en l'absence d'instructions spéciales, les médicaments doivent être dissous dans un glucose de 5% de R-RE-RE (PH 3.5-6.5), une solution isotonique NaCl (pH 4.5-7.0 ).
La solution de glucose, un HCl stabilisé, incompatible avec adrénaline, benzylpénicilline, apomorphine, canamicine, vitamine C, oléenne, glycosides cardiaques. Les glycosides cardiaques sont incompatibles avec l'atropine, la papaverine, la platinilline. AB est incompatible avec l'héparine, l'hydrocortisone. Les vitamines des groupes sont incompatibles les unes avec les autres, avec des vitamines PP, C. La vitamine RR et avec entre elles sont incompatibles.
Il est impossible de mélanger d'autres médicaments: phénothiazide, chlorpromazine, barbituriques, préparations de vitamine C, amphotéricine, furosémide, sunfadiazine, aminophylline, adrénomimétique.
Types de pharmacothérapie. Problèmes déontologiques de la pharmacothérapie.
1. Éthiotrope FT - Correction et élimination des causes de la maladie (AB dans des maladies infectieuses) 2. Pathogenetic FT - Impact sur le mécanisme de développement de la maladie (inhibiteurs ... 3. FT symptomatique - Élimination des symptômes de la maladie quand impossible d'influencer sa cause ou sa pathogenèse (AINS ...Les principes de base du traitement et de la prévention de l'intoxication médicinale. Antidote Thérapie (exemples).
Classification des substances d'empoisonnement:
1. En appartenant à certaines classes composants chimiques: Barbiturats, benzodiazépines, cyanures.
2. D'origine: Nébiologie Nature (acide, alcalis, sels de métaux lourds), produits toxiques d'activité vitale de certains MB (botulinumoxine), origine de légumes (alcolaoïdes, glycosides), origine animale (poisons serpents, abeilles)
3. En degré de toxicité: a) extrêmement toxique (DL50< 1 мг/кг) б) высоко токсические (1-50) в) сильно токсические (50-500) г) умеренно токсические (500-5000) д) мало токсические (5000-15000) е) практически нетоксические (> 15.000)
4. Selon des mesures toxicologiques: a) Neuro-paralytique (bronchospasme, hachage) B) Skin-résorbatif c) général-toxique (crampes hypoxiques, coma, paralysie) d) étouffement d) étouffement d) larme et ennuyant e) psychotrope (violation du mental activité, conscience)
5. Selon la sphère d'utilisation préférentielle: poisons industriels, pesticides, poisons ménagers, substances d'intoxication combattantes, substances médicinales.
6. En fonction de la toxicité du médicament: la liste des médicaments, la nomination, l'utilisation, le dosage et le stockage de ceux dus à une toxicité élevée doivent être effectués avec une prudence élevée. La même liste comprend les médicaments causant une toxicomanie; Liste de B - HP, but, application, dosage et stockage dont il convient de faire des précautions en relation avec complications possibles Lorsqu'il est appliqué sans contrôle médical.
L'effet toxique sélectivement des médicaments.
a) Cardiotoxic: glycosides cardiaques, préparations de potassium, antidépresseurs
b) Neurotoxique: agents psychopharmacologiques, oxychinolines, aminoglycosides
c) hépatotoxique: tétracyclines, lévomycétine, érythromycine, paracétamol
d) néphrotoxique: vancomycine, aminoglycosides, sulfonamides
e) gastroenterotoxique: fonds anti-inflammatoires de stéroïdes, AINS, Reserpine
e) hématotoxique: cytostatique, lévomycétine, sulfonamides, nitrates, nitrites
g) pneumotoxique
Toxicocinétique - étudie l'aspiration, la distribution, le métabolisme et l'élimination des médicaments adoptés dans des doses toxiques.
L'apport de substances d'empoisonnement dans le corps est possible a) entéral B) parentérale. La rapidité et l'exhaustivité de l'aspiration reflètent le taux de développement de l'effet toxique et de sa gravité.
Distribution dans le corps: VD \u003d D / CMAX est un volume valide dans lequel une substance d'empoisonnement est distribuée dans le corps. VD\u003e 5-10 L / kg est difficile à enlever (antidépresseurs, phénothiazines). Vd.< 1 л/кг – ОВ легче удалить из организма (теофиллин, салицилаты, фенобарбитал).
Surdose - Modifications des processus pharmacocinétiques: solubilité, communications avec des protéines, métabolisme ® Augmentation significative de la fraction libre Effet toxique LS ®.
La cinétique de premier ordre avec une augmentation de la concentration de LS passe dans la cinétique de commande zéro.
Stage toxigène - Thérapie désinfectante, étape somatogène - thérapie symptomatique.
Toxicodynamique . Mécanismes de base toxiques:
a) Médiateur: droit (selon le type de blocus compétitif - Fos, psychomimétiques) et indirects (activateurs ou inhibiteurs d'enzymes)
b) Interaction avec les biomolécules et les structures intracellulaires (substances hémolytiques)
c) métabolisme par type de synthèse mortelle ( éthanol, thiophos)
d) Enzyme (poisons serpents, etc.)
Types d'action: local, réflexe, résorbatif.
Classification de l'empoisonnement:
1. Éthiopathogénétique:
a) aléatoire (auto-traitement, erreur)
b) délibéré (avec le but de suicide, meurtre, développement de l'état impuissant affecté)
2. Clinique:
a) Selon le taux de développement d'intoxication: aigu (admission une fois ou avec un court intervalle de la dose toxique de substance), subaiguë (développement lent image clinique après une réception ponctuelle), chronique
b) En fonction de la manifestation du syndrome principal: la défaite de la Cass, la défaite de DS, etc.
c) En fonction de la gravité de l'état du patient: lumière, moyenne, sévère, extrêmement lourde
3. Nonzologique: prend en compte le nom de la LS, le nom du groupe de substances
Le mécanisme de décès global d'intoxication:
a) la défaite du CSS:
1) diminution de la pression artérielle, de l'hypovolémie navires périphériques, Effondrement, brady ou tachycardie (antidépresseurs tricycliques, bêta-bloquants, bloquants de canal de calcium)
2) Arythmies (tachycardie ventriculaire, fibrillation - antidépresseurs tricycliques, théophylline, amphétamine)
b) défaite du système nerveux central: stupeur, dépression respiratoire Koma ® (médicaments, barbituriques, alcool, hypno-sédatif LS)
c) Crampes, hyperréactivité musculaire et rigidité ® Hyperthermie, Mioglobinurie, insuffisance rénale, hypercalémie
Triade toxicologique:
1) L'historique de la durée de l'application, de la dose et de la substance ®.
2) Évaluation de l'état de conscience dans les symptômes: respiration, pression artérielle, température corporelle
3) Données de laboratoire
Principes de base du traitement:
JE. D'abord soins d'urgence : Souffle artificiel, massage cardiaque, thérapie anti-lente, contrôle de l'équilibre de l'eau et de l'électricité
II. Le délai d'aspiration et l'élimination du corps ne sont pas tentés:
But: arrêter le contact avec
1. Chemin parentérale:
a) à travers les poumons:
1) arrêter l'inhalation
2) substances irritantes (ammoniac, formaldéhyde) ® Fixez les mouvements actifs, au chaud, donnez de l'oxygène et des dessinateurs (de alcool ammona Vinaigre de mousse et formaldéhyde - Solution diluée d'alcool d'ammoniac)
b) à travers la peau: laver avec beaucoup de eau chaude Avec du savon ou du détergent, antidote spécifique, neutralisation et résiliation des effets de l'OB sur la peau (FOS: laver à l'eau, enlevé avec 10-15% de l'alcool ammonique ou une solution d'hydrocarbonate de sodium à 5-6% avec de l'eau; laminage de phénol : huile végétale ou éthylène glycol, mais il est impossible huile de vaseline, KMNO 4: 0.5-1% Solution acide ascorbique ou des volumes égaux de 3% de peroxyde d'hydrogène et de solution à 3% acide acétique, CCL 4, Stakidar, essence: chaud eau savonneuse)
c) Lorsque des injections dans la limbe: harnais au-dessus du site d'injection
d) En entrant dans l'oeil: lavage avec une solution saline ou du lait chaud pendant 10 à 20 minutes, pour creuser un anesthésique local; Si les acides et les alcalis, il est impossible de neutraliser. La consultation d'un ophtalmologiste est requise.
2. Chemin entéral: libérer l'estomac de Oh, accélérer le passage
a) élimination de s:
1) Pre-Prendre de l'eau. Il est impossible de prendre du lait (exception - des substances d'intoxication caustiques) et de l'éthanol (exclusion -metthanol).
2) Vomissements - est présenté principalement en cas d'empoisonnement avec de grandes pilules ou de capsules qui ne peuvent pas passer à travers la sonde. Vous pouvez provoquer réflexivement ou vomi (NaCl: 1 cuillère à soupe pour 1 tasse d'eau; Sirop d'hyquaan: adulte 2 cuillères à soupe, enfants 2 cuillères à café; moutarde: 1-2 cuillères à café sur un verre d'eau; Apomorphine: 5-10 mg / kg sous-cutané sauf les enfants de moins de 5 ans). Il est impossible de provoquer des vomissements après la réception: solvants biologiques - Le danger des inhalations, des détergents - Mousse, convulsives RY - Le danger de l'aspiration, des substances caustiques - des dégâts œsophagiens)
3) Protéger l'estomac - est un événement d'urgence et obligatoire. L'estomac est lavé, sinon plus de 4 à 6 heures passa du moment d'empoisonnement, parfois jusqu'à 10 heures; Avec empoisonnement l'acide acétylsalicylique - Après 24 heures. Pré-intuber le patient avec un tuyau avec un brassard gonflable: dans un état comateux en l'absence d'une toux et d'un réflexe laryngé. L'estomac est lavé à l'eau ou à 50 ° C physiathique, l'heure de la procédure est de 4 heures ou plus. À la fin du charbon activé par lavage et du sulfate de sodium.
b) Réduction de l'aspiration de l'aspiration gastro-intestinale: carbone activé à l'intérieur après la vidange de l'estomac + du sulfate de sodium ou du magnésium. Caractéristiques des mesures pour réduire l'aspiration:
1) solvants organiques: il est impossible d'induire des vomissements, de laver l'estomac après l'intubation, du carbone actif + de l'huile de vaseline
2) détergents: Vous ne pouvez pas causer de vomissements et rincer l'estomac, il est nécessaire de donner beaucoup d'eau + de dégoamers (symétiques)
3) acides et alcalis: il est impossible de provoquer des vomissements, de laver l'estomac à travers la sonde, lubrifiée huile végétale Après l'introduction d'analgésiques narcotiques, c'est la seule indication du lait de don. En empoisonnement acide - antiacides, avec empoisonnement alcalin - acide citrique ou acétique.
III. Retrait du corps d'un bain de chaux
a) Dirésis forcés (conditions: flux sanguin rénal suffisant et filtrage glomère; en 24 heures pour verser 20-25 litres)
b) hémodialyse péritonéale
c) Hémosorption
d) Transfusion d'échanges de sang
e) hyperventilation forcée
Iv. Thérapie symptomatique des troubles fonctionnels.
2) toxicocinétique - accélérer la biotransformation OB (bromure de trimpédoxy, thiosulfate de sodium, éthanol, jsc) 3) pharmacologique - atropine, naloxone 4) antidote immunologiqueRègles d'écriture d'énergie toxique, narcotique et puissante.
Recettes pour Narcotic V-VA (morphine, obanopon, promodol, fénamine, cocaïne, etc.) et leur équivalent, indépendamment de LEK. F-Nous écrivons à des promotions. ... dans le cas de LEK. CP-en contenu. L'alcool, alors le timbre est dit. Créatif "Pour ... dans les cas où max. dose de streaming toxique ou streaming. B-C dépasse, éventuellement. Spécifiez leur quantité dans les mots d'ajout ...Ce que nous ferons avec le matériel obtenu:
Si ce matériel s'est avéré utile pour vous, vous pouvez le sauvegarder sur votre page de réseau social:
Des informations sur le moment de l'aspiration, de la distribution et de l'élimination, c'est-à-dire que la pharmacocinétique des substances médicinales peut être exprimée mathématiquement. Ceci est nécessaire lors de la planification des régimes cliniques préparations médicinales. Sur la base des données pharmacocinétiques, les principes de sélection rationnelle et de dosage de ces derniers sont en cours de développement. Dans le même temps, avec ces calculs, un contrôle clinique constant sur l'action du médicament est nécessaire, car les études pharmacocinétiques ne complètent que ce contrôle et vous permettent de tirer des conclusions plus objectives.
L'élimination de la plupart des substances de léopage se produit conformément à la cinétique exponentielle, à savoir, de telle manière que pour chaque période égale, une partie constante de la quantité totale de la substance médicamenteuse injectée disparaît. Dans la plupart des cas, le taux de disparition de la substance médicinale du corps est reflété dans le taux approprié de réduction du niveau du médicament dans le plasma.
La concentration de médicaments dans les fluides biologiques est déterminée par le procédé de chromatographie liquide ou de chromatographie en phase radio, radioimmunale ou enzymatique, polarographie ou spectrophotométrique. La redétermination des concentrations du médicament dans le sang au cours du traitement est appelée surveillance thérapeutique. À cette fin, la salive est parfois utilisée, ce qui est un sang ultrafiltrate de crevettes.
Sur la base des valeurs obtenues, une planification est construite sur l'axe Abscisse dont le temps d'échantillonnage est noté et la concentration du médicament dans l'échantillon biologique (le plus souvent dans le plasma sanguin) dans les unités concernées. La courbe résultante caractérise les processus pharmacocinétiques survenant avec le médicament. Ainsi, après une introduction ponctuelle, la concentration de la substance médicamenteuse dans le plasma diminue de manière exponentielle. Le taux du processus exponentiel peut être caractérisé via une constante de vitesse (K), reflétant le changement de concentration par unité de temps ou après la période du processus séquentiel (indiqué comme t 1/2 ou T / 2). Cette période est égale au temps nécessaire pour terminer le processus de 50%.
La suppression des médicaments de l'organisme peut être jugée par une période de demi-vie ou de semi-émission, semi-émission, qui est déterminée comme le moment de réduire la concentration du médicament dans le sang de 50% de la quantité injectée du médicament ou la suppression de 50% de la quantité biodisponible du médicament.
Le terme «période de semi-élimination» a plus de succès que la période de «demi-vie», car les médicaments ne sont pas seulement affichés, mais également la biotransform. La période de semi-élimination peut être déterminée en fonction de la planification "concentration", mesurant l'intervalle de temps pour lequel une concentration de la substance sur la courbe a diminué de moitié.
Il est presque important de se rappeler que dans une période de demi-vie
50% du médicament est dérivé de l'organe, en deux périodes - 75%, en trois périodes - 90%, en quatre à 94%.
Étant donné que, pour une élimination complète du type exponentiel, une période est nécessaire plus de quatre (4) périodes demi-vie, puis avec une réintroduction du médicament, un cumul (accumulation) est noté pendant des intervalles plus courts. On estime que pour atteindre un plateau de concentration, c'est-à-dire une concentration constante du médicament dans le plasma nécessite environ quatre périodes de demi-vie biologique de la drogue.
Il est important qu'une diminution de l'élimination de la drogue conduit à l'élimination de la période biologique de la demi-vie et à la prolongation du médicament.
Dans certains médicaments, l'action pharmacologique peut être plus longue que nécessaire sur la base de leur T / 2. À cet égard, des médicaments tels que l'hormone de croissance, l'anapriline peut être administré avec des intervalles plus longs que leur T / 2.
Afin d'éviter une augmentation dangereuse du niveau de la drogue dans le plasma chez les patients présentant une élimination réduite en violation du système de foie, de la fonction rénale, du rein ou du système cardiovasculaire, il est nécessaire de réduire les doses de maintenance soit en réduisant chaque dose, soit à l'allongement des intervalles entre l'administration proportionnellement à l'allongement de la période de leur demi-vie biologique.
Accessibilité biologique des médicaments
Pour rendre l'effet thérapeutique, la substance médicamenteuse doit être livrée à ces organes ou tissus dans lesquels son effet spécifique (dans la biofase) est effectué. Avec une administration intravasculaire, le médicament tombe immédiatement et complètement dans le sang. Sous d'autres manières d'administration (oralement, in / m, p / k, etc.) Avant d'entrer dans la circulation sanguine, la substance médicamenteuse doit passer un certain nombre de membranes cellulaires biologiques (mucosa de l'estomac, cellules hépatiques, muscles, etc.) et seulement alors Une partie apparaît dans le flux sanguin systémique. L'effet du médicament dépend en grande partie de la partie de la dose administrée du médicament entre un flux sanguin systémique. Cet indicateur caractérise l'accessibilité biologique des moyens (F). Ainsi, le corps, la biodisponibilité du médicament reflète sa concentration des récepteurs, c'est-à-dire dans le sang et les tissus du corps après une aspiration. Naturellement, la biodisponibilité des mêmes moyens sera différente chez chaque patient. Il est évident qu'avec l'administration des intrants du médicament, la biodisponibilité de celui-ci est d'environ 100% et avec d'autres modes d'administration, la biodisponibilité n'atteint presque jamais à 100%.
Distinguer la biodisponibilité absolue et relative. La biodisponibilité absolue est la proportion de la drogue absorbée lors d'une administration d'imagerie par rapport à son numéro après une administration vénique.
Un indicateur important est la biodisponibilité relative, qui détermine le degré d'absorption relative du médicament de la préparation des tests et des médicaments de comparaison. En d'autres termes, la biodisponibilité relative est déterminée pour divers épisodes de médicaments, pour les médicaments lorsqu'ils sont traités
technologie de production pour les préparations libérées par divers fabricants pour diverses formes posologiques. Pour déterminer la biodisponibilité relative, les données sur le niveau de substance médicamenteuse dans le sang peuvent être utilisées ou son excrétion avec l'urine après une administration unique ou multiple. Ce terme est important lors de la comparaison de 2 médicaments entre eux.
La biodisponibilité comparative des mêmes médicaments fabriquées par différentes entreprises (un exemple: de divertissement polonais-kocarboxinase et fabriquée à Dnepropetrovsk) est déterminée en comparant des équivalences chimiques, biologiques et thérapeutiques.
L'équivalence chimique est une coïncidence des médicaments non seulement formule chimique médicaments, mais aussi une coïncidence de l'isomérisme, la configuration spatiale des atomes dans la molécule de la substance médicamenteuse.
L'équivalence biologique signifie une concentration égale substance active Dans le sang en prenant le médicament de différentes entreprises.
Enfin, l'équivalence thérapeutique implique le même effet thérapeutique équivalent.
Si la liste énumérée 3 caractéristiques coïncide, on dit que les préparations de médicaments ont une biodisponibilité égale (biodisponible). Actuellement, il existe de nombreux exemples du fait que des médicaments similaires sont biologiquement non possibles en raison de différences de biodisponibilité. Le praticien doit se souvenir de cela, surtout lors du transfert d'un patient avec un médicament sur drogue similaire Une autre entreprise.
Il est définitivement que seules les nouvelles sciences peuvent répondre à toutes ces questions - à savoir la pharmacologie clinique. Ceci est une science indépendante avec son sujet et ses objectifs de l'étude. Pourquoi s'est-elle démarquée dans un sujet indépendant? Tout d'abord, car, comme il s'est avéré, tout le monde ne peut pas être étudié dans une expérience animale. Par example, processus mentauxqui sont extrêmement caractéristiques d'une personne seulement.
Le développement rapide de l'industrie pharmaceutique a conduit à la création d'une énorme quantité de médicaments. Avalanche est apparu, créant une jungle médicinale particulière. La situation actuelle rend très difficile de choisir signifie nécessaire Même dans un groupe de médicaments, le médecin empêche le médecin de l'outil optimal d'un patient particulier. La pharmacologie clinique aide à répondre à toutes ces questions.
À titre d'exemple, il est possible d'obtenir la possibilité de choisir un médicament dans les colmamines (maladies du tissu conjonctif, arthrite rhumatoïde, rhumatisme, lupus rouge systémique, etc.). Avec un
les parties sont de l'acide acétylsalicylique (aspirine), mais dans le même temps, il existe d'autres analgésiques non nocifs modernes, comparés à l'aspirine, à un nombre d'avantages: naproxène, pyroxie, etc.
Quoi de mieux, quel médicament ce patient sera plus adéquat, ce qui donne l'effet thérapeutique le plus prononcé? La pharmacologie clinique aide à répondre à ces questions.
Les principales tâches du pharmacologue clinique sont les suivantes:
1) Le choix des médicaments pour le traitement de la douleur concrète
2) Définition du médicament le plus approprié
formes et modes de leur utilisation.
3) Le choix du chemin de l'administration du médicament.
4) Surveiller l'observation de l'action du médicament.
À cette fin, il existe des capteurs qui donnent une image constante de la concentration du médicament dans le sang sur le moniteur. Tous les autres aspects de la pharmacocinétique sont étudiés.
5) L'étude des réactions indésirables et des effets secondaires sur la drogue, leur élimination, ainsi que l'étude des effets de l'interaction des médicaments chez ce patient.
6) Transfert de connaissances accumulées en apprenant.
7) Organisation du laboratoire et services d'informationainsi que des conseils de planification de la recherche (OMS, 1971).
Pharmacodynamique (FD) est une section d'études de pharmacologie
1) Mécanismes d'action (c'est-à-dire l'essence des procédés d'interaction avec les tissus, les récepteurs cellulaires ou de sous-couche - spécifiques ou non spécifiques) 1.
1 02) Effets pharmacologiques (c'est-à-dire le contenu et le changement du médicament, selon l'âge, le sexe du patient, la nature et le flux de la maladie, la pathologie concomitante), ainsi que 3) la localisation des médicaments. Une FD plus courte peut être définie comme une section de pharmacologie qui étudie l'effet des médicaments sur le corps.
Typiquement, le mécanisme de l'action du médicament est étudié dans des expériences animales, car ils sont presque toujours les mêmes chez les animaux et les humains. La connaissance du mécanisme de l'action du médicament permet au médecin de choisir intelligemment le médicament souhaité pour le traitement.
Les mécanismes de l'action des médicaments sont beaucoup, mais tous peuvent être réduits en 2 groupes.
Le premier groupe de mécanismes est associé à ces cas lorsque les médicaments agissent sur des récepteurs spécifiques - c'est-à-dire que ce sont des mécanismes de récepteur.
Le deuxième groupe de mécanismes est associé à des médicaments dus à leur propriétés physico-chimiques Agir non par des récepteurs. Ici, tout d'abord, vous pouvez spécifier l'effet des médicaments sur les enzymes spécifiques de leur effet physico-chimique sur les membranes cellulaires et l'interaction chimique directe avec les substances cellulaires.
À titre d'exemple de mécanismes de non-points, vous pouvez apporter
affaire avec des médicaments pour anesthésie, disons avec le fluorotan. C'est un excellent solvant de graisses, donc tout d'abord des actes sur les membranes des cellules nerveuses, provoquant un effet pharmacologique - anesthésie.
Nous analyserons les mécanismes de récepteurs principaux et les plus souvent trouvés.
Les récepteurs des termes pharmacologiques sont des structures de membrane macromoléculaire biochimiques fonctionnelles, sensiblement sensible à l'action de certains composés chimiques et dans notre cas à l'action des médicaments. Recherche dernières années a montré que les récepteurs pharmacologiques sont des protéines ou des enzymes (protéines G - une chaîne peptidique unique de 7 domaines) - dans cette différence fondamentale des récepteurs morphologiques.
La sensibilité électorale du médicament au récepteur signifie que le médicament peut, tout d'abord, contacter le récepteur, c'est-à-dire qu'il a une affinité ou une affinité pour cela. En d'autres termes, une affinité ou une affinité signifie la capacité du médicament à communiquer avec le récepteur.
Affinité ou affinité reflète les constantes cinétiques reliant la substance médicinale, le récepteur et la réaction à niveau moléculaire. L'interaction des substances médicinales avec le récepteur conduit à un certain nombre de biochimiques et changements physiologiques Dans le corps, qui sont exprimés dans un effet particulier.
La deuxième caractéristique de la substance médicinale est la capacité de causer une réponse pharmacologique, l'effet après l'interaction avec le récepteur. Cette capacité est désignée comme l'activité interne du médicament ou de son efficacité. Dans une certaine mesure, la réaction biologique est régulée en modifiant le nombre de récepteurs et de leur sensibilité.
Dans le processus d'évolution, un récepteur sensible à divers régulateurs endogènes ont été formés. Selon la théorie des récepteurs, le mécanisme de l'action des médicaments consiste à modifier la vitesse de fonctionnement de systèmes d'organisme spécifiques lorsqu'il est exposé à des médiateurs naturels ou à des substances exogènes pour les récepteurs.
Médicaments dont l'action est associée à une excitation directe ou en augmentant fonctionnalité Les récepteurs (capacités) sont appelés agonistes et substances qui empêchent l'action d'agonistes spécifiques, d'antagonistes. En d'autres termes, si la substance médicamenteuse a toutes deux caractéristiques (c'est-à-dire une activité affinité et interne), alors c'est un agoniste. Par conséquent, l'agoniste est une substance avec une grande affinité au récepteur et à une activité interne élevée. Si la substance n'a la capacité que de se lier au récepteur (c'est-à-dire qu'elle a une affinité), mais en même temps incapable de causer des effets pharmacologiques, il provoque le blocus du récepteur et s'appelle un antagoniste.
Préparations qui ont la même affinité pour le récepteur comme agoniste ou plus faible, mais possédant un interne moins prononcé
activité, appelée agonistes partiels ou agoniste antagoniste. Ces médicaments utilisés simultanément avec des agonistes réduisent l'effet de ces derniers en raison de leur capacité à occuper le récepteur.
Exemple: Atropine - a une activité supérieure à celle de l'acétylcholine (médiateur endogène). L'atropine prévaut avec les récepteurs, mais comme il n'a pas d'activité interne, l'effet physiologique ne cause pas. Compte tenu de la plus grande affinité pour le récepteur par rapport à l'acétylcholine, il empêchera l'action de l'agoniste, à savoir l'acétylcholine et donc son antagoniste.
Les substances médicinales peuvent agir comme ou opposées aux médiateurs endogènes. Si la substance médicamenteuse agit comme un médiateur (acétylcholine, noraderennyline, etc.), une telle substance est appelée mimétique. MIM est la racine "MIME", Pantomime, Mimurry. D'où la cholinomimétique, les adrénomimétiques.
Une substance médicinale empêchant l'interaction du médiateur avec un récepteur est appelée bloqueur (cholinoblocator, adrénoblocator, l'histaginoblocator, etc.).
Dans la littérature, vous pouvez rencontrer le terme "litik" (lyse - dissolution, processus physique). Le terme est assez vieux, mais parfois utilisé (cholinolitique, adrénolitique). Ainsi, les termes "litik" et "bloqueur" sont utilisés comme synonymes.
En pratique médicale de plus en plus grande application Trouve le but simultané de plusieurs médicaments. Dans le même temps, ils peuvent interagir les uns avec les autres, modifier la gravité et le caractère de l'effet de base, de sa durée ou de l'affaiblissement des influences latérales et toxiques. À cet égard, la section spéciale de la pharmacodynamique est consacrée à l'interaction des médicaments, classée comme suit. Interaction pharmacologique et interaction pharmaceutique sont isolées.
L'interaction pharmaceutique est associée à l'incompatibilité pharmaceutique des médicaments dans le processus de fabrication ou de stockage, ainsi que lors du mélange dans une seringue. Dans ce cas, l'activité pharmacologique antérieure de médicaments diminue ou disparaît, et parfois même de nouvelles propriétés toxiques apparaissent.
L'interaction pharmacologique des médicaments est associée à des changements dans leur pharmacocinétique, leur pharmacodynamique ou basé sur une interaction chimique et physicochimique dans les médias du corps. Dans le même temps, les médicaments peuvent interagir les uns avec les autres à n'importe quel moment de les transmettre à travers le corps du patient: pendant la succion, dans la phase de transport, dans le processus de métabolisme, ainsi que de l'excrétion (interaction pharmacocinétique).
L'interaction pharmacodynamique reflète le changement des processus associés à chaque médicament individuellement lié à la mise en œuvre de l'effet. En d'autres termes, le type d'interaction pharmacodynamique repose sur les particularités des changements dans les mécanismes et la localisation de l'utilisation de médicaments utilisés, leurs principaux effets. Si l'interaction est effectuée au niveau du récepteur, elle concerne principalement les agonistes et les antagonistes de divers types de récepteurs. Dans ce cas, une substance médicamenteuse peut améliorer ou relâcher l'effet de l'autre. Si les vaisseaux médicinaux
les sociétés agissent en relation avec l'effet de l'unidirectionnelle - ce sont des synergistes de drogue (péché, ergo - travail). Ainsi, la synergie est accompagnée du renforcement de l'effet final. En règle générale, ces substances médicinales agissent sur les mêmes récepteurs. Mettez en surbrillance 2 options de synergie:
1) Les effets coïncident sur le principe d'une quantité simple. Résumée (ou additif, lat. - Additio - Ajouter). L'effet est observé avec un simple ajout des effets de chacun des composants. Par exemple, donc interagir pour l'anesthésie (azote + fluorotan). Semblable à un effet additif avec une utilisation simultanée de l'aspirine et de l'analgin. Pourquoi avez-vous besoin de savoir? Si l'aspirine du patient est obligé de prendre pendant longtempsIl est nécessaire de prendre en compte que l'aspirine agit à l'Ulzerogeniquement, c'est-à-dire qu'elle provoque l'ulcération de la membrane muqueuse du tractus, et l'analgine a un effet aussi indésirable que l'oppression de la formation de sang. Compte tenu de l'effet analgésique additif, il est possible sans un risque substantiel de son occurrence de réduire, de réduire considérablement les dosages des deux outils pris par les patients.
2) La deuxième version des synergies est la potentialisation ou l'effet de l'effet. Cette option se produit lorsque lorsque deux substances sont introduites, l'effet global dépasse la quantité d'effets des deux moyens. À titre d'exemple, l'interaction des neuroleptiques (aminazine) et des médicaments pour anesthésie, l'interaction des antibiotiques et des sulfonylamides antimicrobiens peuvent être obtenues.
Parfois, la troisième (3) variante de synergie est distinguée, - sensibilisation. Sensibilisation - Lorsqu'un médicament dans la dose minimale améliore l'effet d'un autre dans leur combinaison (l'utilisation de petites doses d'insuline en combinaison avec KCL augmente le niveau de pénétration de potassium dans les cellules).
En plus des synergies, il y a un phénomène antagonisme. La capacité d'une substance unique à un degré ou une autre pour réduire l'effet d'un autre est appelée antagonisme, c'est-à-dire un médicament empêchant l'autre.
Mettre en évidence l'antagonisme physique, chimique et physiologique. Cette espèce L'interaction est la plus souvent utilisée dans l'empoisonnement de la surdose ou de la drogue aiguë. Un exemple d'antagonisme physique peut indiquer la capacité des outils d'adsorbant pour faire une aspiration de substances de tube digestif (Adsorbant du carbone activé sur son poison de surface; cholestyramine).
L'illustration de l'interaction chimique peut être la formation de complexes (ions de métaux lourds - mercure, plomb - lie la pénicillamine, EDTA), ou donc interagit acide hydrochlorique Bicarbonate d'estomac et de sodium (alcalin).
L'antagonisme physiologique est associé à l'interaction des médicaments au niveau du récepteur, dont le caractère a déjà été mentionné ci-dessus.
Par analogie avec synergie, il est droit de distinguer (lorsque les deux composés de drogue agissent sur les mêmes récepteurs) et indirects (localisation différente de l'action des médicaments) antagonisme. À son tour, l'antagonisme direct est compétitif et non
COMPÉTITIF. Avec l'antagonisme concurrentiel, la substance médicinale entre dans des relations concurrentielles avec des régulateurs naturels (médiateurs) pour des sites contraignants dans des récepteurs spécifiques. Le blocus du récepteur causé par un antagoniste compétitif peut être éliminé par de grandes doses d'une substance agoniste ou d'un médiateur naturel.
L'antagonisme non concurrentiel est la situation où la substance médicinale peut déplacer le médiateur naturel du récepteur, mais forme des liens covalents avec elle (médiateur).
Points d'interaction de la drogue principale
la masse de récepteurs est située sur l'extérieur et à l'intérieur de la membrane cellulaire et de ses organites. Les points d'interaction les plus fréquents de LS comprennent: 1) les médiateurs et les récepteurs hormonaux; 2) Pompe NA / K Phase ATP, CA, K et Na-NA-PRÉACHTÉS.
Ce dernier prouve une nouvelle fois que la LANS agit sur les mécanismes clés disponibles des réactions borogiques, c'est-à-dire des processus déterministes phoénétiquement, et non en créant de nouvelles réactions.
L'interaction de LS avec le récepteur se produit au niveau des processus chimiques ou physicochimiques. Le plus souvent la nature de la réaction, sa force, sa réversibilité et sa durée est due aux propriétés de la liaison LS avec un récepteur. La force de la communication dépend de la distance de l'interaction électrostatique entre deux atomes. En règle générale, la nature de l'interaction est compliquée, elle peut y participer différentes sortes La communication, qui est déterminée par la complémentarité du réseau local et le récepteur, le degré de convergence d'eux.
Les obligations faibles sont des Vanderwals (déterminez la spécificité de l'interaction des substances avec des systèmes réactifs). Dans la plupart des cas, des liaisons d'ions (réversibles) se produisent entre les médicaments et le récepteur.
Types d'action de la drogue
1) L'action locale est l'effet d'une substance qui se produit sur le lieu de son application. Exemple: l'utilisation de l'anesthésiques locaux est l'introduction d'une solution de Dicain à la cavité de la conjonctive. En utilisant une solution de novocaïne de 1% lors de l'extraction
dent. Ce terme (action locale) est quelque peu conditionnel, car une action véritablement locale est extrêmement rare, car car les substances peuvent être partiellement absorbées ou pour avoir une action réflexe.
2) L'action Reflex est lorsque la substance médicinale agit sur les chemins réflexes, c'est-à-dire qu'elle affecte l'extero ou l'intérieur et les effets et l'effet est manifesté en changeant l'état ou en correspondant aux centres nerveux ou aux organes exécutifs. Ainsi, l'utilisation de morceaux de moutarde dans la pathologie des organes respiratoires améliore leur trophique réflexe (l'huile de moustache essentielle stimule les extérieurs de la peau). Le médicament cytithon (analptique respiratoire) a un effet excitant sur les chimiorétoles du planeur carotidien et, stimulant réflexe le centre respiratoire, augmente le volume et la fréquence de la respiration. Un autre exemple est l'utilisation de l'alcool ammonique dans l'évanouissement (ammoniac), améliore de manière réflexe
circulation cérébrale et centres de vie tonifiants.
3) Un effet gastronomique est lorsque l'effet de la substance se développe après son aspiration (résorption - aspiration; LAT. - RESORBEO - I ARDV), revenu dans la circulation sanguine globale, puis en tissu. L'effet résorbatif dépend des voies d'administration du médicament et de sa capacité à pénétrer dans les barrières biologiques. Si la substance interagit uniquement avec un récepteur de récepteurs à une limite fonctionnel d'une certaine localisation et n'affecte pas les autres récepteurs, l'effet d'une telle substance est appelé sélectif. Ainsi, certaines substances ressemblant à des bandes (relaxants musculaires) bloquent plutôt sélectivement les cholinorécepteurs des plaques de terminaux, provoquant une relaxation des muscles squelettiques. L'effet de la prazosine est associé à un effet d'adrénorécepteurs alpha-un postynaptique postynaptique sélectif et postynaptique qui conduit en fin de compte à réduire la pression artérielle. La base de l'élection de la LS (sélectivité) est l'affinité (affinité) de la substance au récepteur, qui est déterminée par la présence dans la molécule de ces substances de certains groupes fonctionnels et l'organisation structurelle générale de la substance la plus adéquate Interagir avec les récepteurs de données, c'est-à-dire complémentaires.
Caractéristiques générales de l'action des médicaments sur le corps
Malgré l'abondance de médicaments, toutes les impressions,
causés par eux dans le corps, ont une certaine communauté et
envie de dormir. Basé sur le concept du taux de réaction, distinguer 5
types de changements
moyens pharmacologiques (N. V. Verhinin):
1) tonifier (augmenter la fonction à la normale);
2) excitation (améliore la fonction au-dessus de la norme);
3) action apaisante (sédatif), c'est-à-dire une diminution fonction accrue À la normale;
4) inhibition (réduction de la fonction inférieure à la normale);
5) Paralysie (résiliation de la fonction). La quantité de tonification et de chariot
effets de vie appelé action.
Les principaux effets de la drogue
Tout d'abord, distinguer:
1) Effets physiologiques Lorsque des médicaments provoquent de tels changements qu'une augmentation ou une diminution de la pression artérielle, de la fréquence cardiaque, etc.
2) biochimique (augmentation des enzymes sanguines, glucu
shl, etc.). De plus, allouer principal (ou principal) et
Nezernaya (mineures) effets de la drogue. EF de base.
L'effet est celui sur lequel le médecin construit ses calculs dans le traitement de ce (!) Patient (analgésique - pour un effet anesthésique, hypotensant - pour réduire la pression artérielle, etc.).
Effets non fondamentaux ou non-indispensables, contraire, ceux qui sont inhérents ce milieuMais le développement de ce patient est facultatif (analgésique non narcotique - en plus de l'effet analgésique, l'effet antipyrétique est causé par l'effet antipyrétique, etc.). Parmi les effets non essentiels, il peut y avoir des effets désirables et indésirables (ou côté).
Exemple. Atropine - détend les muscles lisses de l'interne
organes. Cependant, en même temps, il améliore simultanément la conductivité dans le cœur de l'auto (sous le blocus du cœur), augmente le diamètre de l'élève, etc. Tous ces effets doivent être considérés individuellement dans chaque cas particulier.
Facteurs affectant l'ampleur de l'effet de médicaments
1) Tout d'abord, vous devez vous rappeler les facteurs pharmacocinétiques inhérents à chaque médicament. Cela a déjà été mentionné ci-dessus, je ne vous rappelle que surtout que nous parlons de son taux d'aspiration ou de son absorption, une biotransformation, son excrétion (médicament, médicament).
2) Le deuxième groupe de facteurs est physiologique.
a) l'âge. En effet, tout le monde sait que la sensibilité du patient pour la drogue change avec l'âge. Indiqué même à cet égard:
Pharmacologie périnatale;
Pharmacologie pédiatrique;
Pharmacologie gériatrique;
Pharmacologie de la reproduction;
b) la masse du patient. On sait que plus la masse est grande, plus
dose. Par conséquent, la dose de drogue dans (mg / kg).
c) Paul. Une sensibilité différente est détectée chez les hommes et les femmes
à certaines substances, par exemple, à la nicotine, à l'alcool, etc.,
ce qui est expliqué par la différence de métabolisme, la différence de spécifique
poids de la couche de graisse et ainsi de suite
c) l'état du corps. Action ls sur le corps après su
l'exercice public sera différent de rien.
e) Rythmes biologiques (quotidien, mensuel, saisonnier, année
vous, et maintenant même la population) ont le plus grave
l'effet sur l'effet des médicaments dans le corps. 3) faits pathologiques
ry (par exemple, le niveau d'activité hormonale). Alors quand B.
la maladie zeed est plus facile à transporter les doses toxiques de la morphine, mais la sensibilité du myocarde à l'adrénaline augmente. 1 L'effet des glycosides cardiaques sur la circulation sanguine ne se manifeste que sur le fond de l'insuffisance cardiaque. L'effet de LS est considérablement varié dans les hypo-et hyperthermies, avec des maladies infectieuses, avec une modification de l'état fonctionnel du CNS, etc.).
4) Facteurs génétiques. On sait que l'absence de l'enzyme glucose-6-phosphatedhydrogénase (M.) pendant la thabulation, empêche de prescrire des préparations antipaludiques telles que la primahine. L'insuffisance de l'enzyme bootrylcholinestérase dans le sang, se produisant dans l'une des 2500 personnes, est la cause de la minélaxation à long terme pour l'introduction de la dithiline.
5) l'impureté des patients ou des effets de placebo. À cet égard, l'effet anticanalique des médicaments placebo atteint par exemple 40% et jusqu'à 81% de l'effet placebo résulte du trajet d'injection de l'administration de médicaments. Probablement donc utiliser préparations de vitamines, Tonic signifie que les tranquillisants sont en grande partie dues à cet effet.
6) dose de médicaments. L'effet de la drogue est très déterminé par leur dose. Dose appeler la quantité de médicaments
une société destinée à une réception (généralement appelée dose unique). Non seulement l'efficacité du traitement, mais également la sécurité du patient dépend de la dose du médicament. Même à la fin du quartier de Hush Century, William Villas a écrit: «Le poison de petites doses est le meilleur médicament; médicament utile dans trop de dose - poison». C'est le plus correct à notre époque où des médicaments extrêmement actifs sont introduits dans la pratique médicale, dont les doses sont mesurées par les actions d'un milligramme.
Dénoter la dose en grammes ou en grammes. Pour un dosage plus précis de drogues, leur nombre par 1 kg de poids corporel est calculé (ou par mètre carré. M de la zone du corps), par exemple, 1 mg / kg; 1 μg / kg, etc. Le médecin doit être orienté non seulement dans une dose conçue pour une seule réception (pro DOSI), mais également dans une dose quotidienne (PRO DIE).
Les doses minimales dans lesquelles des médicaments provoquent l'effet biologique initial (thérapeutique) sont appelés seuils, ou des doses de manière minimale (thérapeutique). En médecine pratique, les doses thérapeutiques moyennes sont les plus souvent utilisées dans lesquelles des médicaments ont l'effet pharmacothérapeutique optimal nécessaire. Si, avec leur ordonnance, le patient n'est pas suffisamment exprimé, la dose est élevée à la dose thérapeutique la plus élevée. Des doses thérapeutiques plus élevées peuvent être ponctuelles et quotidiennes. La dose ponctuelle la plus élevée est la quantité maximale d'un médicament qui sans préjudice au patient peut être entrée une fois. Ces doses sont rares, dans des cas extrêmes (dans la situation urgente et urgente). Les doses thérapeutiques moyennes sont généralement de 1/3-1 / 2 de la dose unique la plus élevée.
Les doses thérapeutiques les plus élevées de toxique et substances puissantes Ils sont donnés dans la pharmacopée d'État de l'URSS. Dans certains cas, par exemple, lors de l'utilisation des agents chimiothérapeutiques, la dose de la drogue sur le traitement du traitement (dose à terme) est indiquée. S'il devient nécessaire de créer rapidement une concentration élevée du médicament dans le corps (sepsis, échec cardiovasculaire), utilisez la première dose de la dose dite de choc, qui dépasse toutes les personnes suivantes. Il existe également toxique (fournissant des effets dangereux) et des doses meurtrières.
Le médecin est important de connaître une autre caractéristique - à savoir le concept de la latitude de l'effet thérapeutique du médicament. Sous la latitude de l'action thérapeutique, la distance, la distance de la thérapeutique minimale à la dose minimalotoxique est comprise. Naturellement, plus cette distance, plus ce médicament est sécurisé.
1/20 dose x nombre d'années pour enfants
Pour la caractéristique quantitative et l'évaluation de l'efficacité du nouvel agent pharmacologique, deux comparaisons standard sont utilisées, avec un placebo ou avec le médicament analogique
type d'action gychique, qui est l'un des plus efficaces de ce groupe de fonds.
Le placebo (mannequin) est une substance indifférente d'une forme posologique imitant une certaine pharmacologie ou une certaine drogue. L'utilisation de placebo est nécessaire en la présence: a) l'effet de l'estimation, l'impact de l'individu, l'attente et relations biaisées du patient ou du chercheur; b) Des changements spontanés dans le cours de la maladie, des symptômes, ainsi que des phénomènes non liés au traitement.
Placebo - terme latin, signification: "Je peux te livrer plaisir."
L'effet placebo est l'effet causé par des propriétés pharmacodynamiques non spécifiques du médicament avec une pathologie donnée, mais le fait de l'utilisation de médicaments qui affecte psychologiquement. Les préparations de placebo sont généralement inertes pharmacologiquement, elles contiennent des substances inactives similaires à l'amidon ou au lactose. Utilisation de placebo dans des études cliniques afin d'établir l'effet de la suggestion du patient et du médecin, en particulier si l'étude est soumise à des fonds destinés au traitement. l'asthme bronchique, maladie hypertendue, angine d'angine, IHS. Dans de tels cas, le médicament placebo ne doit pas être en couleur et d'autres propriétés physiques (odeur, goût, forme) diffère de la drogue active. Le placebo est plus efficace lorsque le médecin et le patient sont peu informés de lui.
EXEMPLE. Pour maladie ischémique COEURS (IBS) Si un groupe de patients atteints d'IBS prescrit un médicament actif et l'autre placebo, puis 40% des patients atteints du deuxième groupe d'attaques d'angine sont arrêtés.
Les plus effet prononcé Le placebo (jusqu'à 81%) est observé avec un système inexting de l'introduction. Les médicaments et les pilules sont moins efficaces.
Dans la littérature consacrée à l'influence de la toxicomanie sur le patient, le terme pharmacothérapie (FT) sonne souvent. La pharmacothérapie est une section de pharmacologie, étudiant la thérapie avec des médicaments médicinaux.
Distinguer espèce suivante Pharmacothérapie:
1) étiotrope - vue parfaite Pharmacothérapie. Ce type de FT vise à éliminer la cause de la maladie. Des exemples de FT étiotropes peuvent être traités avec des moyens antimicrobiens de patients infectieux (benzylpénicilline dans la pneumonie streptocoque), l'utilisation d'antidéticules dans le traitement des patients atteints d'une intoxication toxique
substances. 2) Pharmacothérapie pathogénétique - Dirigé
Éliminer ou
suppression des mécanismes de développement des maladies. La plupart des médicaments s'appliquent actuellement précisément au groupe de médicaments de la FT pathogénétique. Les agents antihypertenseurs, les glycosides cardiaques, antiarythmiques, anti-inflammatoires, psychotropes et de nombreux autres médicaments ont un effet thérapeutique en supprimant les mécanismes pertinents de développement des maladies.
3) Thérapie symptomatique - visant à éliminer ou
restriction de certaines manifestations de la maladie. Les médicaments symptomatiques comprennent des analgésiques qui n'affectent pas la cause ou le mécanisme pour le développement de la maladie. Les moyens bénéfiques sont également un bon exemple de moyens symptomatiques. Parfois ces moyens (élimination syndrome de hibou avec infarctus du myocarde) peut avoir un impact significatif sur le courant processus pathologique Et en même temps, jouez le rôle des moyens de thérapie pathogénétique.
4) La pharmacothérapie de remplacement est utilisée avec une pénurie de substances biogéniques naturelles. Les préparations enzymatiques (pancréatine, PANZINORM, etc.) comprennent une thérapie de remplacement (PANZINORM, etc.), des médicaments hormonaux (insuline dans le diabète sucré), préparations en vitamines (vitamine D, par exemple avec des rachés). Préparatifs de la thérapie de substitution, sans éliminer les causes de la maladie, peut assurer l'existence normale du corps pendant de nombreuses années. Ce n'est pas par hasard que cette pathologie sévère comme diabète - Il est considéré comme un style de vie spécial chez les Américains.
5) La thérapie préventive est effectuée afin d'empêcher les maladies. Prophylactique comprend certains outils antiviraux (Par exemple, à l'épidémie de grippe - Rematadine), des désinfectants et un certain nombre d'autres. L'utilisation de préparations anti-tuberculose du type isoniside peut également être considérée comme une ft prophylactique. Bon exemple La thérapie préventive est l'utilisation de vaccins.
De la pharmacothérapie doit être distingué par chimiothérapie. Si la FT traite de deux participants dans le processus pathologique, à savoir le médicament et le macroorganisme, alors pendant la chimiothérapie, il y a déjà 3 participants: la médecine, le macroorganisme (patient) et l'agent causatif de la maladie.
En parlant de doses, nous avions d'abord indiqué sur des doses allopathiques, contrairement à la homéopathie. Par conséquent, quelques mots sur l'homéopathie. Le terme "homéopathie" est formé de deux mots grecs: homois est similaire et pathétique - souffrant, maladie. Littéralement, l'homéopathie est traduite comme une telle similarité. Fondateur de l'homéopathie Scientifique allemand Samuel Ganeman dans son célèbre livre "Organe d'art médical ou la principale théorie du traitement homéopathique" début xix. Des siècles (1810) ont décrit les principes de base de cette science. Les principes de ceux-ci sont plusieurs, mais 2 d'entre eux sont la principale:
1) Il s'agit d'une loi similaire, qui stipule que le traitement des maladies doit être effectué par un moyen similaire et similaire. Selon ce principe, Ganeman conseille de "imiter la nature qui guérit parfois la maladie chronique à travers une autre maladie de l'adhésion". Par conséquent, "contre la maladie à guérir (principalement chronique) devrait être utilisée une telle substance médicamenteuse capable de provoquer une autre, celle qui est destinée à une maladie artificielle similaire, et le premier sera guéri." Similia.
sIMILIBUS (similaire à celui-ci). Par exemple, la jaunisse doit être traitée jaune, etc.
2) Le deuxième principe est de traiter des doses supermales. La dilution des médicaments utilisés par les homéopathes est calculée dans plusieurs ordres, atteignant parfois leurs dizaines: 10 en cinquième; 10 dans le dixième; 10 en dix-huitième ou plus (c'est-à-dire des millions
et plus de grammes). Expliquer l'effet d'appliquer le
cartinités B.
des dilutions élevées de Ganeman ont mis en avant un concept spéculatif: "Les petites doses sont caractérisées par une force spirituelle spéciale, une activité accrue, la capacité de pénétrer dans les organes et les tissus affectés."
On ne sait pas à quoi une puissance spirituelle spéciale n'est pas connue, mais la vie scientifique a présenté des preuves très graves au cours de la dernière décennie à la Justice de l'approbation de Haneman. Par exemple, les expériences du Jacquenist français, produites par celle-ci avec une dilution de substances à 10 heures dans un jour de huit jouet, ont montré que les molécules d'eau possèdent une "mémoire" à la présence de cette substance, ce qui provoque un certain effet physiologique. Si le fait a noté que ce fait découvre une confirmation dans un proche avenir, c'est-à-dire si elles établissent si les molécules d'eau ne sont pas une source d'informations, elles seront certainement sur les fondements des plus grandes découvertes qui peuvent expliquer l'efficacité thérapeutique des remèdes homéopathiques.
Ensuite, examinez la section concernant les aspects pharmacologiques de l'effet toxique des médicaments, à savoir la toxicologie des médicaments. La toxicologie des médicaments est la section de la pharmacologie, qui étudie les effets toxiques de ces fonds. Cependant, maintenant, il est plus correct de parler de réactions indésirables Le corps humain pour les médicaments. Ce fait est connu depuis longtemps, le matériau réel riche est accumulé, indiquant que les réactions indésirables divers degrés Peut se produire lors de la prise de presque tous les médicaments.
Il existe de nombreuses classifications des effets secondaires des médicaments et des complications de la pharmacothérapie, bien qu'aucun d'entre eux ne soit parfait. Dans le même temps, sur la base du principe pathogénétique, tous les effets indésirables ou réactions peuvent être divisés en 2 types:
1) les réactions indésirables associées à
a) Surdosage de drogues
b) empoisonnement;
2) Réactions toxiques associées aux propriétés pharmacologiques des médicaments.
Surdosage se produit généralement lors de l'utilisation de fortes doses de médicaments. Surtout, une surdose se produit lors de la prise de médicaments qui ont une petite latitude d'action thérapeutique. Par exemple, des manifestations de toxicité sont difficiles à éviter d'utiliser des antibiotiques d'aminoglycoside (streptomycine, kanamycine, néomycine). Ces médicaments provoquent des troubles vestibulaires et de la surdité lors du traitement des doses qui dépassent ceindiquement thérapeutique. Pour certains médicaments, il est tout simplement impossible d'éviter les complications toxiques (antitumoral, médicaments cytotoxiques), qui endommagent toutes les cellules divisées rapidement et UG
moelle osseuse Netone avec effet simultané sur la croissance des cellules tumorales.
De plus, une surdose peut être associée non seulement à des doses élevées, mais également à un phénomène de cumulement (glycosides coeur).
L'empoisonnement peut être aléatoire et délibéré. L'intoxication intentionnelle se produit généralement avec un objectif suicide (à des fins de suicide). Dans la région Omsk le plus souvent dans structure commune Les performances occupent une intoxication par des fluides migrateurs, à la deuxième place sont une intoxication médicinale. C'est tout d'abord d'empoisonner avec des somnifères, des tranquillisants, des fos, des alcools de monoxyde de carbone.
Malgré la différence de facteurs étiologiques, les mesures d'assistance aux étapes de l'allocation médicale sont imprimées similaires.
Ces principes sont les suivants:
1) Combattre la nade non nominale du tractus gastro-intestinal. Le plus souvent, il est nécessaire pendant un poison d'empoisonnement oral. L'intoxication tranchante la plus fréquente est causée par la réception des substances à l'intérieur. Obligatoire et d'urgence à cet égard est de laver l'estomac à travers la sonde même de 10 à 12 heures après l'empoisonnement. Si le patient est conscient, le lavage de l'estomac est effectué en utilisant grand nombre L'eau et les vomissements ultérieurs causent. Cause de vomissement chemin mécanique. Dans un état inconscient, le lavage de l'estomac du patient est effectué à travers la sonde. Il est nécessaire d'envoyer un effort à l'adsorption dans l'estomac du poison, pour lequel le charbon actif est utilisé (1 cuillère à soupe à l'intérieur ou 20-30 comprimés en même temps, avant et après la lavage de l'estomac). L'estomac est lavé plusieurs fois 3-4 heures jusqu'à la purification complète de la substance.
Les vomissements sont contre-indiqués dans les cas suivants:
Au cours des états comateux;
En empoisonnant avec des liquides corrosifs;
En cas de kérosène, d'une intoxication à l'essence (possibilité de pneumonie hydrocarbonate avec nécrose tissu pulmonaire etc.).
Si la victime est un petit enfant, il est préférable d'utiliser pour le lavage solutions En petits volumes (100-150 ml).
De l'intestin, le poison est mieux éliminé à l'aide de laxatifs salés. Par conséquent, à la fin du lavage, vous pouvez entrer dans un estomac de 100 à 150 ml d'une solution de sulfate de sodium à 30% et encore plus de sulfate de magnésium. Les laxatifs de sel sont les plus puissants et fonctionnent rapidement dans tout l'intestin. Leur action obéit les lois de l'osmose, de sorte qu'elles couvrent l'effet du poison pendant une courte période.
C'est bon de donner astringent (solutions de tanin, de thé, de cerisier), ainsi que de l'enveloppant (lait, protéines d'œufs, huile végétale).
Lorsque le poison est expulsé, il est nécessaire de rincer complètement la peau, la meilleure eau de liège. En cas de substances toxiques, il devrait arrêter leur inhalation à travers les poumons, en supprimant les affectés de l'atmosphère empoisonnée.
Avec l'administration sous-cutanée de la substance toxique, l'aspiration du lieu d'administration peut être ralentie par les injections de la solution d'adrénaline.
autour du lieu d'introduction de la substance, ainsi que le refroidissement de cette zone (glace sur la peau sur le site d'injection).
2) Le deuxième principe d'assistance dans une intoxication aigu est d'effet sur le poison d'étanchéité, l'élimination de celui-ci du corps.
Dans le but de l'élimination précoce de la substance toxique du corps, appliquez principalement des diurines forcées. L'essence de cette méthode consiste à combiner la charge d'eau améliorée avec l'introduction de diurrent actifs et puissants. L'inondation du corps est effectuée par abondante Patient ou introduction à / dans différentes solutions (solutions absorbant le sang, glucose, etc.). Du diurétique, le plus souvent utilisé furosemid (Laziks) ou un mannitol. Nous aimons les tissus du patient "rinçage" telle qu'elle était, libérant de la substance toxique. Cette méthode ne peut supprimer que les substances libres qui ne sont pas liées à des protéines et aux lipides sanguins. Devrait prendre en compte la balance des électrolytes qui, lorsqu'elles sont utilisées cette méthode Il peut être perturbé en raison de l'élimination d'un nombre important d'ions du corps.
Dans une insuffisance cardiovasculaire aiguë, une violation prononcée de la fonction rénale et du danger du développement de l'œdème cérébral ou des poumons, la diurèse forcée contre-indiquée.
Outre la diurée forcée, l'hémodialyse et la dialyse péritonéale sont utilisées, lorsque le sang (hémodialyse ou rein artificiel) traverse une membrane semi-perméable, libérant des substances toxiques ou un "lavage" de la cavité du péritoine avec une solution d'électrolytes est effectué.
Méthodes de désintoxication extracorporelle. Une méthode de détoxification réussie qui a été répandue est la méthode d'hémosorption (lymphosorption). Dans ce cas, les substances toxiques du sang sont adsorbées sur des sorbants spéciaux (charbon granulé recouvert de protéines sanguines, allenizenka). Cette méthode permet de détoxifier avec succès l'organisme en empoisonnement avec des neuroleptiques, des tranquillisants, des Fos, etc. La méthode d'hémosorption est des substances dérivées mal éliminées par hémodialyse et dialyse péritonéale.
Utilisez la substitution sanguine lorsqu'elle est combinée de la sangle avec une transfusion de sang donneur.
3) Le troisième principe de lutte contre l'intoxication aigu est de neutraliser le poison Vostely en introduisant des antagonistes et des antidotes.
Les antagonistes sont largement utilisés dans une intoxication tranchante. Par exemple, atropine en empoisonnement avec des agents anticholinestérase, FOS; Nallane - avec empoisonnement de morphine, etc. Les antagonistes pharmacologiques interagissent généralement de manière compétitive avec les mêmes récepteurs que des substances qui ont provoqué une intoxication. À cet égard, il semble très intéressant de créer des anticorps spécifiques (monoclonaux) par rapport aux substances, qui sont particulièrement souvent la cause d'une intoxication tranchante (anticorps monoclonaux contre glycosides cardiaques).
Pour le traitement spécifique des patients présentant des produits chimiques avec des produits chimiques, la thérapie antidigment est efficace. Les antidotes appellent des fonds utilisés pour une liaison spécifique de poison, neutra
lisser, inactivant des poisons ou par interaction chimique ou physique.
Ainsi, pendant une intoxication, des métaux lourds utilisent des composés qui forment des complexes non toxiques avec eux (par exemple, Unitiol avec empoisonnement arsenic, le Dr Penicylamine, Reforthell avec empoisonnement avec des préparations de fer, etc.).
4) Le quatrième principe consiste à mener une thérapie symptomatique. Spécial grande importance La thérapie symptomatique acquiert des substances dans une intoxication qui ne disposent pas d'antidotes spéciaux.
La thérapie symptomatique soutient les fonctions essentielles: la circulation sanguine et la respiration. Glycosides coeur d'occasion, Vasotonic, agents qui améliorent la microcirculation, la thérapie à l'oxygène, les stimulants respiratoires. Les causes sont éliminées par les injections de Sibazon. À l'œdème du cerveau, la thérapie de déshydratation (furosémide, mannite) est effectuée. Les analgésiques sont utilisés, l'état du sang acide-alcalin est corrigé. Lorsque vous arrêtez, la respiration est transférée au patient sur ventilation artificielle Poumons avec un complexe de mesures de réanimation.
Ensuite, nous nous concentrerons sur le deuxième type de réactions indésirables, c'est-à-dire des réactions indésirables associées aux propriétés pharmacologiques des médicaments. Les effets secondaires des médicaments se manifestent chez 10 à 20% des patients ambulatoires et 0, 5 à 5% des patients ont besoin d'une hospitalisation pour corriger les troubles de HP. Celles-ci indésirables, en termes de pathogenèse, des réactions peuvent être: a) droit et b) associé à la sensibilité modifiée du corps du patient.
Nous analyserons des réactions toxiques directes. Ils sont appelés droit parce que les médicaments directement directement toxiquement système fonctionnel. Par exemple, un antibiotique aminoglycosidal (streptomycine, kanamycine, gentamicine) expose neurotoxicité, ayant un effet toxique sur le corps auditif (outtoxy) et l'appareil vestibulaire. en plus cette classe Les antibiotiques ont une toxicité pour les réactions comportementales qui se manifestent par la léthargie, l'apapathie, l'intensité, la somnolence.
Les médicaments peuvent avoir des réactions gélatotoxiques directes. Par exemple, le fluorotan (médicament pour anesthésie) lors de ses applications répétées dans court instant Il peut avoir un effet toxique prononcé jusqu'à la dystrophie de foie jaune aiguë.
Les effets toxiques directs peuvent être réalisés par néphrotoxicité. L'effet des antibiotiques micolin-aminoglycosides est possédé. Lors de la prescription des préparations de cette série, le patient nécessite une surveillance constante des tests d'urine (protéines, sang dans l'urine, etc.).
L'effet toxique droit suivant est l'ulutérine (ulcérateur). Par exemple, la nomination de salicylate, de glucocorticoïdes, de moyens antihypertensens de reserpine, conduit à l'ulcération de la muqueuse de l'estomac, qui doit être prise en compte lors de la nomination de ces classes de fonds, en particulier des patients souffrant déjà d'une maladie ulcéreuse.
Les effets toxiques directs peuvent être exprimés en embryotoxicité. Laissez-moi vous rappeler que l'embryotoxique s'appelle défavorable
l'effet des médicaments qui ne sont pas associés à une violation de l'organogenèse, découlant jusqu'à 12 semaines de grossesse. Et l'effet toxique des médicaments dans une période de grossesse ultérieure s'appelle féotoxique. Il est nécessaire de se rappeler cet effet lors de la prescription des médicaments aux femmes enceintes, de la conduite de la pharmacothérapie par des témoignages stricts.
Exemples: 1) Le but de la streptomycine enceinte peut entraîner une sourde au fœtus (lésion VIII paire d'encrachant et nerfs cérébraux); 2) Les tétracycles affectent négativement le développement des os du fœtus; 3) La mère souffrant de morphinetisme, le nouveau-né peut également souffrir de dépendance physique à la morphine.
Les médicaments peuvent avoir une tératogénicité, c'est-à-dire une telle influence dommageable sur la différenciation des tissus et des cellules, ce qui conduit à la naissance des enfants avec des anomalies différentes. Par exemple, l'utilisation de thalidomide, qui a un effet tératogène prononcé, qui a un effet tératogène prononcé, conduit à la naissance dans des pays Europe de l'Ouest Plusieurs milliers d'enfants avec diverses déformations (membres de la Focomelia - Lust-like; Amélie - la disparition des membres; Hémangions de la personne, anomalies gastro-intestinales).
Pour étudier les effets tératogènes des substances, l'influence des médicaments sur les animaux étudie, bien qu'il n'y ait aucune corrélation directe sur les effets des substances médicinales sur les animaux et une personne. Par exemple, dans la même tératogénicité de la thalidomide dans l'expérience de souris a été révélée à une dose de 250 à 500 mg / kg de masse, et il s'est avéré être égal à 1-2 mg / kg.
Le premier trimestre est le plus dangereux pour l'action tératogène (en particulier une période de 3 à 8 semaines de grossesse), c'est-à-dire une période d'organogenèse. Dans ces temps, il est particulièrement facile de provoquer une lourde anomalie pour le développement de l'embryon.
Lors de la création de nouveaux médicaments, il est également nécessaire d'avoir la possibilité d'effets négatifs graves tels que la mutagénicité chimique et la cancérogénicité. La mutagnation est la capacité des substances à causer des dommages persistants à la cellule germinale, mais en particulier de son appareil génétique, qui se manifeste dans le changement du génotype de progéniture. La cancérogénicité est la capacité des substances à provoquer un développement tumeurs malignes. Les estrages contribuent au développement du cancer du sein chez les femmes en âge de procréer.
Les effets mutagènes et tératogènes peuvent se manifester des mois et même des années, ce qui rend difficile l'identification de leur véritable activité. La tératogénicité est inhérente aux agents antinéoplasiques, corticostéroïdes, androgènes, alcool. Cyclophosphamide, certains agents hormonaux ont un effet cancérogène.
Les réactions indésirables lors de l'utilisation de médicaments peuvent être exprimées par le développement de la toxicomanie ou si une dépendance à une drogue plus globale. Il existe plusieurs signes de base de toxicomanie.
1) Ceci est la présence de la dépendance mentale, c'est-à-dire un tel état lorsque le patient développe une attraction mentale non colonie pour la réinsertion d'une substance médicamenteuse, par exemple
drogue.
2) Dépendance physique - ce terme dénote la présence d'une grave indisposition physique chez un patient sans réinjection de la substance médicamenteuse, en particulier de drogues. Avec une nette arrêt de l'administration du médicament qui a causé la dépendance à la drogue, le phénomène de privation ou d'abstinence se développe. La peur semble, anxiété, aspirant, insomnie. Peut-être l'anxiété moteur, l'agressivité se produit. De nombreuses fonctions physiologiques sont violées. Dans des cas graves, l'abstinence peut être la cause du décès.
3) Le développement de la tolérance, c'est-à-dire une dépendance. Autres types
les effets indésirables causés par les propriétés des médicaments eux-mêmes sont des troubles associés aux changements dans le système immunobiologique du patient lors de la réception de médicaments très actifs. Par exemple, l'utilisation d'un large éventail d'antibiotiques d'action peut se manifester avec une modification de la flore bactérienne normale du corps (intestins), mise en œuvre par le développement de la superinfection, la dysbactériose, la candidose. Le plus souvent, les poumons et les intestins sont impliqués dans ces processus.
La corticostérothérapothérapie et la thérapie immunosuppressive affaiblissent l'immunité, entraînant un risque de développement maladies infectieusesTout d'abord, nature opportuniste (pneumocysteose, cytomégalovirus, etc.).
Ce sous-groupe de réactions est de 2 types:
1) réactions allergiques;
2) idiosyncrasie. Il faut dire que négatif
associé à
les réactions communes se trouvent très souvent dans la pratique médicale. Leur fréquence augmente tout le temps. Ils se présentent indépendamment de la dose du médicament injecté et des mécanismes immunisés sont impliqués dans leur formation. Les réactions allergiques peuvent être 2 espèces: l'hypersensibilité du type immédiat, le GNT - associé à la formation d'anticorps de classes IgE et IgG4) et une lente (accumulation de t-lymphocytes t-lymphocytes et de macrophapes et de macrophages).
La photo clinique est très diversifiée: urticaire, éruption cutanée, œdème de l'eurhème, maladie sérique, asthme bronchique, fièvre, hépatite, etc. Mais la principale chose est la possibilité de développer un choc anaphylactique. Si au moins deux fois le contact du patient avec une substance médicamenteuse est nécessaire pour le développement de réactions allergiques, le développement de l'idiosyncraysie - intolérance des substances médicinales pendant le contact primaire avec xénobiotique est toujours associé à un défaut génétique, en règle générale, absence ou activité extrêmement faible de l'enzyme. Par exemple, l'utilisation de la préparation anti-antitimalarium de primahine chez les personnes ayant une enzymopathie génétique (insuffisance de la loi. M. G-6-FDG) provoque la formation de Quinone, qui a une action hémolytique. En présence de cette fermentation, il est dangereux pour la cession de médicaments qui sont des oxydants, car cela peut conduire à une hémolyse
Érythrocytes, anémie hémolytique médicinale (aspirine, levomycétine, comté, primahine, furadonine).
Quelques mots sur la création de nouveaux médicaments, évaluer les médicaments et leur nomenclature. Les progrès de la pharmacologie se caractérisent par une recherche continue et la création de nouveaux médicaments. Créer des médicaments commence par la recherche de chimistes et de pharmacologues, dont la coopération créative est absolument nécessaire lors de l'ouverture de nouveaux médicaments. Dans le même temps, la recherche de nouveaux fonds se développe dans plusieurs directions.
La manière principale est la synthèse chimique des médicaments, qui peut être réalisée comme synthèse directionnelle ou avoir un chemin empirique. Si une synthèse directionnelle est associée à la reproduction de substances biogéniques (insuline, adrénaline, noréfinerénaline), la création d'antimétabolites (pabk-sulfonamides), modification des molécules de composés avec une activité biologique connue (changement de la structure de l'acétylcholine - Gongliobiledor Hygros) , etc., alors le chemin empirique est soit des découvertes aléatoires, soit une recherche par dépistage, c'est-à-dire de taper divers composés chimiques dans une activité pharmacologique.
On peut donner un exemple de constatations empiriques à réformer un effet hypoglycémique lors de l'utilisation de sulfanimamides, qui ont ensuite conduit à la création d'agents antidiabrames perfoiques synthétiques de sulfonamine (butamide, chloropramide).
Temps très fastidieux et une autre version du chemin empirique de la création de médicaments - méthode de dépistage. Cependant, il est inévitable, surtout s'il est examiné nouvelle classe Les composés chimiques dont les propriétés, basées sur leur structure, sont difficiles à prédire (chemin inefficace). Et ici un grand rôle En cours de lecture de la recherche scientifique de l'informatisation.
À l'heure actuelle, les médicaments sont principalement obtenus par la synthèse chimique directionnelle, qui peut être effectuée a) par similitude (introduisant des chaînes supplémentaires, des radicaux) B) par complémentarité, c'est-à-dire que le respect des récepteurs et des organes de tissu.
Dans l'arsenal des médicaments, en plus des préparations synthétiques, des préparations et des substances individuelles de matières premières médicinales d'origine végétale ou animale sont occupées, ainsi que de divers minéraux. Ce sont principalement Galenov, novogalen, alcaloïdes, glycosides. Donc, de l'opium est obtenu de la morphine, de la codéine, de la Papaverine, de la Rauflphie du Serpentic - Reserpine, d'un flutter - Coeur Glycosides - Digitoxine, Digoxine; À partir d'un certain nombre de roches endocrines de bétail - hormones, préparations immunoactives (insuline, thyroïdine, accident vasculaire cérébral, etc.).
Certains médicaments sont des produits de la vie des champignons et des micro-organismes. Exemple - antibiotiques. Les substances médicinales de légumes, d'origine animale, microbienne et fongiques servent souvent de base à leur synthèse, ainsi que de transformations chimiques ultérieures et d'obtenir des préparations semi-synthétiques et synthétiques.
Digurer le rythme de créer des médicaments en utilisant
méthodes de génie génétique génétique (insuline, etc.).
Un nouveau médicament, passant par tous ces "Sita" (l'étude de la pharmacie, de la pharmacodynamie, de la pharmacocinétique, de l'étude des effets secondaires, de la toxicité, etc.) est autorisé pour les essais cliniques. Il utilise la méthode "Contrôle des aveugles", l'effet placebo, la méthode de double "contrôle aveugle", lorsque ni le médecin ni le patient ne sait lorsque ce placebo est utilisé. Il ne connaît que la commission spéciale. Des essais cliniques sont menés chez l'homme et dans de nombreux pays, il est effectué sur des volontaires. Ici, bien sûr, il existe une masse d'aspects juridiques, déontologiques et moraux du problème qui nécessitent leur développement clair, leur réglementation et leur approbation des lois sur ce compte.
Nomenclature des médicaments
De nombreux médicaments constitués d'une substance active peuvent être nommés en fonction de leur structure chimique. Mais en raison de la grande complexité de leur mémorisation et de l'inconvénient de l'utilisation de noms chimiques dans la pratique médicale ne sont pas utilisés.
Actuellement, 2 types de titres sont utilisés pour désigner des médicaments:
1) international non exclusif, approuvé par les autorités sanitaires officielles et sont utilisés dans les pharmacopes nationaux et internationaux;
2) noms commerciaux ou marqués qui sont la propriété commerciale de Farmfirm. Dans ce cas, le même médicament peut avoir de nombreux noms. Diazen Stranquilizer a des noms de société "Sedusen", "Sibazon", "Relanium", etc. Certains LANs ont plus de 100 articles (par exemple, vitamine B12). Habituellement, sur l'emballage du médicament, il y a un nom de marque et un nom international non propriétaire.
Préféré est la décharge de médicaments sous leurs noms non exclusifs, ce qui réduit la possibilité erreurs médicales. Ces médicaments sont moins chers que les médicaments avec le nom d'entreprise. En outre, la décharge du médicament sous leur nom non spécifique donne à la pharmacie de fournir un patient au médicament de toute entreprise produisant ce médicament.
Pour les cliniciens, la classification la plus pratique des médicaments est celle qui repose sur le principe nosologique (par exemple, des moyens pour le traitement de l'asthme bronchique, de l'infarctus du myocarde, des préparations antidiabétiques, etc.). Mais meilleures classifications prendre en compte de tels signes de drogues telles que la localisation de l'action, l'effet pharmacologique, application thérapeutique. L'une de ces classifications qui sont les plus parfaites est la classification de l'académicien M. D. Mashkovsky, selon lequel son livre de référence bien connu est indiqué.
1. Le concept de traitement comme correction directionnelle des troubles physiologiques dans le corps. Utilisez et risque lorsque vous utilisez des médicaments. Motifs de leur application. Évaluation de sécurité.
Pharmacologie - base théorique de la pharmacothérapie.
Raisons de l'utilisation de médicaments:
1) Pour la correction et l'élimination de la cause de la maladie
2) Si l'insuffisance préventive
3) sur des indications de vie
4) besoin évident, basé sur le niveau de connaissance et l'expérience
5) Le désir d'améliorer la qualité de la vie
Utilisez lors de la prescription des médicaments:
1) Correction ou élimination de la cause de la maladie
2) Faciliter les symptômes de la maladie lorsqu'il est impossible de traiter
3) la substitution de médicaments BAV naturels qui ne sont pas produits par des organismes en quantités suffisantes
4) Mise en œuvre de la prévention des maladies (vaccin, etc.)
Risque - la probabilité que le résultat de l'impact soit un préjudice ou des dommages; Il est égal au ratio du nombre d'événements défavorables (pervers) au groupe de risque.
A) inacceptable (préjudice\u003e bénéfice)
B) acceptable (avantage\u003e préjudice)
C) insignifiant (105 - niveau de sécurité)
D) conscient
L'évaluation de la sécurité du réseau local commence au niveau laboratoires chimiquesSynthétisant des médicaments. L'évaluation préclinique de la sécurité de LS est effectuée par le ministère de la Santé, de la FDA, etc. Si les LANs transmettent avec succès cette étape, son évaluation clinique commence, composée de quatre phases: i Phase - une évaluation de la tolérance sur des volontaires sains 20-25 ans, II - sur des patients atteints de bénévoles de moins de 100 personnes souffrant d'une maladie de certaines maladies, phase III - études cliniques multicentrales dans Grands groupes de personnes (jusqu'à 1000 personnes), phase IV - surveillant le médicament pendant 5 ans après sa permission officielle. Si LS passe avec succès toutes ces phases, elle est considérée comme sûre.
2. L'essence de la pharmacologie en tant que science. Sections et zones de pharmacologie moderne. Les principaux termes et concepts de pharmacologie sont l'activité pharmacologique, l'action et l'efficacité des produits chimiques.
Pharmacologie - Science des médicaments dans tous les aspects - Base théorique de la thérapie:
A) Science sur l'interaction des produits chimiques avec des systèmes vivants
B) Science de la gestion des processus de l'activité essentielle du corps à l'aide de produits chimiques.
Sections de pharmacologie moderne:
1) Pharmacodynamique - études a) l'impact des médicaments sur le corps humain, b) l'interaction de divers médicaments dans le corps tout en les nominant simultanément, c) l'influence de l'âge et de diverses maladies sur l'action de LS
2) Pharmacocinétique - étudie l'aspiration, la distribution, le métabolisme et l'excrétion des médicaments (c'est-à-dire que le corps du patient réagit à la drogue)
3) Pharmacogénétique- étudie le rôle des facteurs génétiques dans la formation de la réponse pharmacologique du corps sur le réseau local
4) Pharmacoéconomique- évalue les résultats de l'utilisation et du coût des médicaments pour prendre une décision sur l'application pratique ultérieure.
5) Pharmacoépideyologie - LS étudie l'utilisation de médicaments et de leurs effets au niveau des populations ou de grands groupes de personnes pour assurer l'utilisation des ls les plus efficaces et les plus sûrs.
Activité pharmacologique (biologique) - la propriété de la substance pour causer des changements dans le biosystème (corps humain). Substances pharmacologiques \u003d substances biologiquement actives (BAV)
effet pharmacologique - l'effet des médicaments sur l'objet et sa cible
Effet pharmacologique - Le résultat de l'action d'une substance dans le corps (modification des processus physiologiques, biochimiques, structures morphologiques) est un changement quantitatif, mais pas qualitatif de l'état des biosystèmes (cellules, tissus, organes).
Efficacité de LS. - La capacité du réseau local à causer certains effets pharmacologiques dans ce cas dans ce cas. Il est estimé sur la base de «preuves substantielles» - des études et des essais cliniques bien contrôlés adéquats effectués par des experts avec une formation scientifique et une expérience scientifique pertinentes pour étudier les médicaments de ce type (FDA)
3. Nature chimique des médicaments. Facteurs fournissant l'effet thérapeutique des médicaments - effet pharmacologique et effets de placebo.
Les agences sont 1) Légumes 2) Animal 3) Microbien 4) Mineral 5) Synthétique
Les LS synthétiques sont présentés par presque toutes les classes de composés chimiques.
effet pharmacologique - l'effet des médicaments sur l'objet et sa cible.
Placebo - Toute composante thérapie qui n'a pas d'impact biologique spécifique sur la maladie, qui fait l'objet de traitement.
Il est utilisé pour contrôler lors de l'évaluation de l'action des médicaments et de profiter au patient sans aucun moyen pharmacologique à la suite d'un impact psychologique (c'est-à-dire Effet placebo).
Tous les types de traitement ont un composant psychologique ou offrent une satisfaction ( Effet placebo), ou causant une anxiété ( Effet nocebo). Un exemple d'effet placebo: amélioration rapide d'un patient avec une infection virale lors de l'utilisation d'antibiotiques qui n'affectent pas les virus.
L'effet de placebo favorable est lié à l'impact psychologique sur le patient. Ce sera le maximum que lorsqu'il sera utilisé En combinaison avec des méthodes de traitementavoir un effet spécifique prononcé. Substances coûteuses Comme un placebo contribue également à la réalisation d'une plus grande réponse.
Indication pour placebo:
1) Troubles mentaux faibles
2) soutien psychologique du patient avec une maladie chronique incurable ou soupçonnée d'un diagnostic important
4. Sources et étapes de la création de médicaments. Définition des concepts Substance médicinale, médecine, médicament et posologie. Nom des médicaments.
Sources de création de médicaments:
A) Matières premières naturelles: plantes, animaux, minéraux, etc. (glycosides coeur, insuline de porc)
B) BAV naturel modifié
C) Composés synthétiques
D) Produits d'ingénierie génétique (insuline recombinante, interférons)
Étapes de la création de médicaments:
1. Synthèse de LS dans le laboratoire chimique
2. Évaluation préclinique de l'activité et des effets indésirables du ministère de la Santé et des autres. Organisations
3. Tests cliniques LS (pour plus de détails, voir. 1)
Médicament - toute substance ou produit utilisé pour modifier ou explorer des systèmes physiologiques ou des conditions pathologiques pour la prestation des prestations (OMS, 1966); Substances individuelles, mélanges de substances ou composition de la composition inconnue, qui ont des propriétés thérapeutiques éprouvées.
Substance médicinale - Composé chimique individuel utilisé comme médicament.
Formulaire de dosage - Confortable pour une application pratique, la forme attachée au médicament pour obtenir l'effet thérapeutique ou prophylactique nécessaire.
Traitement médical - médicament à une certaine formulaire de dosage, autorisé par le gouvernement.
5. Manières d'introduire des médicaments dans le corps et leurs caractéristiques. Élimination de la pression des médicaments.
1. Pour l'action du système
MAIS. Voie d'administration entérale: oralement, sublingual, transbuccifal, rectation, à travers la sonde
B. Route d'administration parentérale: intraveineuse, par voie sous-cutanée, intramusculaire, inhalation, sous-arachnoïde, transdermique
2. Pour une exposition locale: Cauducted (Épicient), sur la muqueuse, dans la cavité (abdominale, pleurale, articulaire), en tissu (infiltration)
|
Chemin d'injection HP |
Dignité |
désavantages |
|
Oralement - à travers la bouche |
1. Pratique et simple pour le patient 2. Aucune stérilité des médicaments |
1. L'absorption de nombreux médicaments dépend du repas, de l'état fonctionnel du tractus et d'autres facteurs, avec des difficultés à prendre en compte dans la pratique. 2. Tous les HS ne sont pas bien absorbés dans le tractus gastro-intestinal 3. Certains ls sont détruits dans l'estomac (insuline, pénicilline) 4. Une partie des pls a une NLR sur la membrane muqueuse gastro-intestinale (AINS - l'expansion des membranes muqueuses, des antiacides - supprimer la moto) 5. Non applicable chez les patients inconscients et en violation de la déglutition |
|
Sublingual et transbukkalo |
1. Administration pratique et rapide 2. Absorption rapide LS 3. LAN n'est pas soumis à une élimination préservée 4. L'effet du médicament peut être rapidement interrompu. |
1. L'inconvénient créé par une utilisation régulière fréquente des comprimés 2. Irritation de la membrane muqueuse de la bouche, une séparation excédentaire de la salive, favorisant l'ingestion de drogues et une diminution de son efficacité 3. goût désagréable |
|
Recto |
1. La moitié de la LV n'est pas soumise à un métabolisme préservé 2. n'irrite pas la digestion muqueuse 3. Idéalement, lorsque d'autres modes d'administration sont inacceptables (vomissements, mal de mer, enfants de la poitrine) 4. action locale |
1. Moments psychologiques désagréables pour le patient 2. L'absorption des médicaments est considérablement ralentie dans un rectum non parlé. |
|
Intravascularité (généralement intraveineuse |
1. Arrivée rapide dans le sang (états urgents) 2. Création rapide de la concentration élevée du système et il est possible de le gérer 3. Vous permet d'entrer dans un LS qui s'est effondré dans le tractus gastro-intestinal |
1. Difficultés techniques d'accès intravasculaire 2. Risque d'infection dans le site d'injection 3. Thrombose des veines sur le site d'injection du LS (érythromycine) et péniblement (chlorure de potassium) 4. Certains ls sont adsorbés sur les murs des gouttes (insuline) |
|
Intramusculaire |
Aspiration assez rapide du médicament dans le sang (10-30 minutes) |
Le risque de complications locales |
|
Sous-cutané |
1. Le patient peut faire des injections indépendamment après la formation. 2. Effet long de ls |
1. Aspiration lente et manifestation de l'effet du réseau local 2. Atrophie de tissu adipeux sur le site d'injection et réduire le taux d'aspiration des médicaments |
|
Inhalation |
1. Départ rapide Actions I. haute concentration Au lieu d'administration dans le traitement des maladies de la respiration. sentiers 2. Bonne surveillance de l'action 3. Réduire les effets du système toxiques |
1. Le besoin d'un dispositif spécial (inhalateur) 2. La complexité d'utiliser des aérosols sous pression pour certains patients |
|
LS local. |
1. Concentration active élevée de drogues sur le lieu d'administration 2. Effets systémiques indésirables de ce réseau local |
En cas d'intégrité altérée de la peau, le médicament peut tomber dans la circulation sanguine systémique - la manifestation des effets systémiques indésirables. |
Élimination de la pression des médicaments (l'effet du premier passage) - le processus de biotransformation du médicament jusqu'à ce que la LS frappe dans le flux sanguin systémique. Dans l'élimination préservée lors de l'administration orale du médicament, les systèmes enzymatiques des intestins, de la veine du sang et des hépatocytes et des hépatocytes sont impliqués.
Avec une administration intraveineuse, une élimination préservée est absente.
Pour oralement reçu par le réseau local action utile, Il est nécessaire d'augmenter sa dose pour compenser les pertes.
6. Transfert de médicaments par des barrières biologiques et ses variétés. Les principaux facteurs affectant le transfert de médicaments dans le corps.
Méthodes d'absorption (transport) dans les membranes biologiques:
1) Filtration (diffusion aqueuse) - mouvement passive des molécules de la substance en fonction du gradient de concentration à travers les pores remplis d'eau de la membrane de chaque cellule et entre les cellules adjacentes est caractéristique de l'eau, des ions, de petites molécules hydrophiles (urée ).
2) La diffusion passive (diffusion lipidique) est le mécanisme principal du transfert de LV, le processus de dissolution du médicament dans les lipides à la membrane et le mouvement à travers eux.
3) Transport à l'aide de supports spécifiques - Le transfert de LV à l'aide des porteurs intégrés à la membrane (plus souvent) est caractéristique des molécules polaires hydrophiles, un certain nombre d'ions inorganiques, de sucres, d'acides aminés, de pyrimidines:
a) Diffusion légère - est réalisée sous un gradient de concentration sans coûts de l'ATP
b) Transport actif - contre un gradient de concentration avec les coûts de l'ATP
Le processus saturable est que le taux d'absorption n'augmente que jusqu'à ce que la quantité de molécules de médicaments soit égale au nombre de supports.
4) Endocytose et Pinocytose - Le médicament est associé à une composante membrane cellulaire reconnaissant spéciale, l'invagoration de la membrane se produit et une bulle contenant la molécule de médicament est formée. Par la suite, le médicament est distingué d'une bulle dans une cellule ou transportée d'une cellule. Caractérisé par des polypeptides de poids moléculaire élevé.
Facteurs affectant le transfert de médicaments dans le corps:
1) Propriétés physico-chimiques de la matière (hydroélectricité et lipophile, ionisation, polarisation, taille des molécules, concentration)
2) Structure des barrières de transfert
3) Blessences
7. Transférer à travers les membranes des substances médicinales avec une ionisation variable (l'équation d'ionisation de Genderson-Gasselbalch). Principes de contrôle de transfert.
Tous les LS sont des acides faibles ou des terrains faibles ayant leurs significations de la constante d'ionisation (RK). Si la valeur polaire du milieu est égale à la valeur du médicament RK, 50% de ses molécules seront sous ionisées et 50% dans l'état non ionisé et le milieu du médicament sera neutre.
Dans un environnement acide (pH inférieur à celui de RK), où il y a un excès de protons, l'acide faible sera de la forme injuste (R-Cooh), c'est-à-dire associé au protoné. Une telle forme acide est non chargée et bien soluble dans les lipides. Si le pH est décalé sur le côté alcalin (c'est-à-dire que le pH deviendra plus que le RK), l'acide commencera à se dissocier et perdra le proton, tout en se transformant en une forme non verte, qui a une charge et est mal soluble dans les lipides.
Dans un milieu alcalin, où il y a un déficit de proton, la base faible sera sous la forme indiscratible (R-NH2), c'est-à-dire que cela sera imprégné et privé de charge. Cette forme de base est bien soluble dans les lipides et est rapidement absorbée. Dans le milieu acide, il y a un excès de protons et une base faible commencera à dissocier, à lier les protons et à former la forme de base chargée protonée. Une telle forme est mal soluble dans les lipides et est mal absorbée.
D'où, L'absorption des acides faibles se déroule principalement dans un environnement acide et des bases faibles en alcaline.
Caractéristiques du métabolisme des acides faibles (SC):
1) Estomac: SK dans la teneur en acide de l'estomac est non ionisée et dans un milieu alcalin de l'intestin grêle, il sera dissocié et les molécules SC acquiert une charge. Par conséquent, l'absorption des acides faibles sera la plus intense de l'estomac.
2) Dans le sang, le milieu est plutôt alcalin et les molécules SK Skidle vont se passer sous forme ionisée. Le filtre des glomers rénaux passe à la fois des molécules ionisées et non ionisées, alors malgré la charge de la molécule, le SC sera émis dans l'urine primaire.
3) Si l'urine est alcaline, l'acide restera sous forme ionisée, ne sera pas capable de les réabsorber dans le sang et de se démarquer de l'urine; L'urine est acide, le médicament passe à une forme non ionisée, qui est facilement réactivée dans le sang.
Caractéristiques du métabolisme des terrains faibles: contraire de manière opposée (l'absorption est meilleure dans les intestins; dans l'urine alcaline soumise à la réabsorption)
T. À propos., Pour accélérer l'élimination du corps de la faible urine acide doit être nettoyée et d'accélérer l'élimination d'une base faible doit être acidifiée (désintoxication par POPOV).
La dépendance quantitative du processus d'ionisation du médicament avec un pH différent du support vous permet d'obtenir l'équation Henderson— Hasselbach.:
Où PKA correspond à la valeur du pH dans laquelle la concentration de formes ionisées et non ionisées est en équilibre .
L'équation de Gasselbach nous permet d'estimer le degré d'ionisation des médicaments à une valeur de pH donnée et de prédire la probabilité de sa pénétration à travers la membrane cellulaire.
(1) Pour diluer acide, a,
HA ↔ H + + A - L'HA est une concentration de formes non ionisées (protonées) de l'acide et une - - la concentration de forme ionisée (non compromée).
(2) pour base faible, b,
BH + ↔ H + + B, où BH + est la concentration de la forme de base protonée, B est une concentration de forme non déplacée
Connaître le pH du milieu et la substance de la PCA peut être déterminée par le logarithme calculé pour déterminer le degré d'ionisation du médicament, et donc le degré de son absorption du tractus gastro-intestinal, de la réabsorption ou de l'excrétion par les reins à différents pH de la urine, etc.
8. Transfert de médicaments dans le corps. Diffusion de l'eau et diffusion dans les lipides (loi FIC). Transport actif.
Le transfert de médicaments dans le corps peut être effectué par la diffusion de l'eau et des lipides, le transport actif, l'endo et la pinocytose.
Caractéristiques Transfert LS dans le corps de la diffusion de l'eau:
1. Couvertures épithéliales (Duals muqueux, cavité buccale, etc.) - Diffusion de l'eau de seules de très petites molécules (méthanol, lithium ions, etc.)
2. Capillaires (sauf pour le cerveau) - filtrage des substances avec du poids moléculaire jusqu'à 20-30 mille oui.
3. Capillaires cérébraux - N'a principalement pas de pores d'eau, à l'exception des zones d'hypophyse, d'épiphyse, de zones de ventriculaire, de plexus choroïdien, d'altitude médiane
4. Placenta - n'a pas de AUMA (bien que la question soit controversée).
5. La liaison des médicaments avec des protéines sanguines empêche leur sortie du sang, et donc la diffusion de l'eau
6. La diffusion dans l'eau dépend de la taille des molécules LS et des pores d'eau
Caractéristiques de la diffusion de lipides:
1. Le mécanisme principal de LS transfère à travers les membranes cellulaires
2. Il est déterminé par la lipophile de la substance diffusable (c.-à-d. Le coefficient de distribution d'huile / eau) et le gradient de concentration peut être limité à une très faible solubilité de la substance dans de l'eau (qui empêche la pénétration des médicaments dans la phase aqueuse de membranes)
3. Connexions non polaires facilement diffuses, ions difficiles.
Toute diffusion (et eau, dans les lipides) obéit la loi de la diffusion fika:
Taux de diffusion - la quantité de molécules de médicaments portables par unité de temps; C1 est la concentration de la substance à l'extérieur de la membrane; C2 est la concentration de la substance de l'intérieur de la membrane.
Conséquence de la loi fika:
1) la filtration LS est plus élevée que sa concentration sur le site d'injection (s absorbée de la surface dans l'intestin plus que dans l'estomac, l'absorption des médicaments dans l'intestin est plus rapide)
2) Le filtrage des faibles est supérieur à la plus grande que la concentration de médicaments sur le site d'injection
3) La filtration LS est supérieure à l'épaisseur la moins épaisseur de la vaste membrane biologique (l'épaisseur de la barrière dans les alvéoles des poumons est nettement inférieure à la peau, de sorte que la vitesse d'absorption est plus élevée dans les poumons)
Transport actif - Transfert LC, quel que soit le dégradé de concentration utilisant l'énergie ATP, est caractéristique des molécules polaires hydrophiles, un certain nombre d'ions inorganiques, de sucres, d'acides aminés, de pyrimidines. Caractérisé: a) Sélectivité à certains composés b) La possibilité de concurrence de deux substances pour un mécanisme de transport B) saturation à fortes concentrations de substances d) la possibilité de transport contre le gradient de concentration d) une énergie considérable.
9. Concentration centrale postule de la pharmacocinétique de médicament sanguin - le paramètre principal pour contrôler l'effet thérapeutique. Tâches résolues sur la base de la connaissance de ce postulat.
Le postulat central (dogme) de la pharmacocinétique: la concentration de LV dans le plasma sanguin détermine (déterminer quantitativement) l'effet pharmacologique.
Dans la plupart des cas, le taux d'absorption, la distribution, le métabolisme et l'excrétion des médicaments sont proportionnels à leur concentration plasmatique sanguine (obéissent la loi des masses actuelles), donc il est possible:
1) Déterminer la période de semi-élimination (pour LS avec cinétique de premier ordre)
2) Expliquez la durée des effets toxiques de certains ls (pour les médicaments à fortes doses avec une cinétique de saturation)
10. BioAvailabilité de la drogue - Définition, essence, expression quantitative, déterminants. Concept de biodisponibilité
BioAvailabilité (F) - caractérise le taux d'exhaustivité et d'absorption des médicaments avec des chemins d'introduction introduits - reflète la quantité de substance inchangée qui a atteint le flux sanguin systémique, par rapport à la dose initiale du médicament.
F est à 100% pour LS, qui sont administrés par voie intraveineuse. Avec l'introduction d'autres moyens, f est généralement moins due à une absorption incomplète et du métabolisme partiel dans les tissus périphériques. F est 0, si le HP n'est pas absorbé par la lumière du tractus gastro-intestinal.
Pour l'estimation F, la courbe de la dépendance de la concentration de médicament dans le sang du temps après son administration par voie intraveineuse, ainsi que après l'administration par l'étude. C'est t. N. Courbes pharmacocinétiques de la dépendance "concentration temporelle". Par intégration, les valeurs de la zone sous la courbe pharmacocinétique sont trouvées et calculées comme relativement:
![]() ≤ 1, où AUC est une zone sous une courbe pharmacocinétique (zone sous la courbe)
≤ 1, où AUC est une zone sous une courbe pharmacocinétique (zone sous la courbe)
BioAvailabilité\u003e 70% est considérée comme élevée, inférieure à 30% - faible.
Déterminants biodisponibilité:
1) Taux d'aspiration
2) Complétisité d'aspiration - une absorption insuffisante du LS en raison de sa très hautes hydrophilie ou de sa lipophile, métabolisme des bactéries intestinales avec administration entérale, etc.
3) Élimination de la pression - avec une biotransformation élevée dans la LS LS LS (nitroglycérine au cours de l'administration orale).
4) forme de dosage - comprimés sublinguaux et suppositoires rectaux Aide à éviter une élimination préventive.
11. Répartition des médicaments dans le corps. Compartiments, ligands. Les principaux déterminants de la distribution.
Distribution LS - Le processus de propagation du réseau local sur les organes et les tissus après avoir entré le flux sanguin systémique.
Compartiments de distribution:
1. Espace extracellulaire (plasma, fluide intercellulaire)
2. Cellules (cytoplasme, membrane organienne)
3. tissu gras et osseux (dépôt LS)
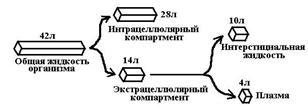 Dans une personne pesant 70 kg, les volumes de milieux liquides sont comme un ensemble de 42 litres, puis si:
Dans une personne pesant 70 kg, les volumes de milieux liquides sont comme un ensemble de 42 litres, puis si:
[VD \u003d 3-4 L, alors tout le médicament est distribué dans le sang;
[VD \u003d 4-14 L, alors tous les médicaments sont distribués dans un fluide extracellulaire;
[VD \u003d 14-42 L, alors tout le médicament est approximativement réparti uniformément distribué dans le corps;
[VD\u003e 42 L, alors tout le médicament est principalement dans l'espace extracellulaire.
Ligands moléculaires LS:
A) récepteurs spécifiques et non spécifiques
B) protéines sanguines (albumine, glycoprotéine) et tissus
C) Polysaccharides Connexion des tissus
D) nucléoprotéis (ADN, ARN)
Distribution des déterminants:
· Nature ls. - Che moins de dimensions Molécules et lipophiles LS, la distribution plus rapide et uniformément.
· Taille de l'organe - Che plus de taille L'Autorité, plus les outils médicinaux peuvent y entrer sans changement significatif dans le gradient de concentration
· Bloodstock dans l'organe - dans les tissus bien perfusés (cerveau, coeur, reins) La concentration thérapeutique de la substance est créée beaucoup plus tôt que dans les tissus pauvres perfusables (graisse, os)
· La présence de barrières histohematiques- LS pénétre facilement le tissu avec une GBB mal prononcée
· Reliure de médicaments avec des protéines plasmatiques - Plus la fraction LS associée est grande, la pire sa distribution dans le tissu, car les molécules libres peuvent quitter le capillaire.
· Dépôt de médicaments dans les tissus - La liaison des médicaments avec des protéines tissulaires contribue à son accumulation, car la concentration de LS gratuite dans l'espace périvasculaire est réduite et un gradient de concentrations élevées entre le sang et les tissus est constamment maintenu.
La caractéristique quantitative de la distribution du médicament est le volume de distribution apparent (VD).
Quantité apparente de distributionVd. - Il s'agit d'un volume hypothétique de fluide dans lequel toute la dose du médicament peut accueillir la concentration à concentrer dans le plasma plasma.
VD est égal à l'attitude de la dose administrée (quantité totale de médicaments dans le corps) à sa concentration dans le plasma sanguin:
 .
.
Plus la quantité apparente de distribution est grande, plus les médicaments sont répartis dans le tissu.
12. Élimination constante, son essence, dimension, connexion avec d'autres paramètres pharmacocinétiques.
Taux d'élimination constante (Kel, min-1) - montre quelle partie du médicament est éliminée du corps par unité de temps þ Kel \u003d AVD / ALCC, où l'avyde est le nombre de médicaments sécrétés dans des unités. Temps, Aubshch - Le nombre total de médicaments dans le corps.
La valeur KEL est généralement découverte en résolvant une équation pharmacocinétique décrivant le processus d'élimination des médicaments du sang, de sorte que Kel s'appelle l'indicateur de modèle de la cinétique. Il n'y a aucune relation directe avec la planification du mode de dosage Kel, mais sa valeur est utilisée pour calculer d'autres paramètres pharmacocinétiques.
La constante d'élimination est directement proportionnelle au jeu et proportionnelle inversement au volume de la distribution (de la définition de la clairance): Kel \u003d CL / VD; \u003d Heure-1 / min - 1 \u003d part par heure.
13. La demi-vie des drogues, son essence, une dimension, une relation avec d'autres paramètres pharmacocinétiques.
Période de semi-élimination (T1, min) est le temps nécessaire pour réduire la concentration de LS dans le sang sans heurts. Dans le même temps, il ne joue pas le rôle de la manière dont la concentration est atteinte - avec l'aide de la biotransformation, de l'excrétion ou de la combinaison des deux processus.
La période de semi-élimination est déterminée par la formule:
![]()
Demi-vie - le paramètre pharmacocinétique le plus important qui permet:
B) déterminer le temps d'élimination complète du médicament
C) prédire la concentration de médicaments à tout moment (pour LS avec cinétique de premier ordre)
14. Dégagement en tant que paramètre principal de la pharmacocinétique pour contrôler le mode de dosage. Son essence, sa dimension et une connexion avec d'autres indicateurs pharmacocinétiques.
Autorisation (Cl, ml / min) - le volume de sang, qui est effacé des médicaments par unité de temps.
T. K. Plasma (sang) est une partie «visible» du volume de distribution, puis de dégagement - fraction du volume de distribution, à partir duquel le médicament est attribué par unité de temps. Si vous désignez la quantité totale de médicaments dans le corps à travers Astrichet le montant qui a été médié par Avyd., ensuite:
![]() D'autre part, de déterminer la quantité de distribution, il s'ensuit que la quantité totale de médicaments dans le corps est Aubsch \u003d.Vd.´
C.Ter / plasma. En substituant cette valeur dans la formule de dédouanement, nous obtiendrons:
D'autre part, de déterminer la quantité de distribution, il s'ensuit que la quantité totale de médicaments dans le corps est Aubsch \u003d.Vd.´
C.Ter / plasma. En substituant cette valeur dans la formule de dédouanement, nous obtiendrons:
![]() .
.
Ainsi, le dégagement est le rapport de la vitesse de la drogue à sa concentration dans le plasma sanguin.
Dans ce formulaire, la formule de dédouanement est utilisée pour calculer la dose de maintenance du médicament ( RÉ.P), c'est-à-dire la dose du médicament qui devrait compenser la perte de médicaments et maintenir son niveau à un niveau constant:
Introduction Vitesse \u003d Taux d'exclusion \u003dCl.´ C.Ter (dose / min)
RÉ.P \u003d Introduction Vitesse´ T. (T. - Intervalle, entre réception de médicaments)
Additif de dégagement, c'est-à-dire que l'élimination de la substance du corps peut survenir avec la participation des processus dans les reins, les poumons, le foie et d'autres organes: Clapochene. + CLAMP + CLRT.
La clairance est connectée Avec une période de semi-élimination des médicaments et de la distribution: T1 / 2 \u003d 0,7 * vd / cl.
15. Dose. Types de doses. Unités de dosage de médicaments. Objectifs de dosage, méthodes et options d'introduction, intervalle d'introduction.
L'effet des médicaments sur le corps est largement déterminé par leur dose.
Dose - la quantité de substance introduite dans l'organisme dans une réception; Il est exprimé en unités de poids, en vrac ou conditionnel (biologique).
Types de doses:
A) dose unique - la quantité de substance pour une réception
B) dose quotidienne - la quantité de médicament assignée à une ou plusieurs réceptions par jour
C) Dose à terme - la quantité totale du médicament pour le traitement
D) Doses thérapeutiques - doses dans lesquelles le médicament est utilisé avec des objectifs thérapeutiques ou préventifs (seuils ou doses thérapeutiques minimales, thérapeutiques et plus élevées).
E) Doses toxiques et mortelles - Doses de LV, dans lesquelles ils commencent à avoir des effets toxiques prononcés ou à causer la mort du corps.
E) la dose de chargement (entrée) - le nombre de LS injecté, qui remplit tout le volume de la distribution du corps dans la concentration active (thérapeutique): DR \u003d (CSS * VD) / F
G) Dose de soutien - Saisie systématique du nombre de médicaments compensant les pertes LS avec dégagement: PD \u003d (CSS * cl * dt) / F
Unités de dosage Pls:
1) dans les grammes ou les fractions de grammes
2) Nombre de médicaments par 1 Kg masses corporelles (par exemple, 1 Mg / kg) ou par unité de surface corporelle (par exemple, 1 Mg / m2.)
Buts de dosage LS:
1) Déterminer le nombre de médicaments nécessaires pour causer l'effet thérapeutique souhaité avec une certaine durée
2) Évitez les phénomènes d'intoxication et d'effets secondaires avec l'introduction de médicaments
Méthodes pour l'introduction de HP: 1) Enteral 2) Parentéral (voir. 5)
Variantes d'administration de la LS.:
A) Continu (par perfusion intravasculaire prolongée ls goutte à goutte ou via des distributeurs automatiques). Avec une introduction continue de médicaments, sa concentration dans le corps varie en douceur et n'est pas soumise à des fluctuations importantes
B) Administration intermittente (méthodes d'injection ou d'injection) - l'introduction de médicaments à certains intervalles (intervalles de dosage). Avec une introduction discontinue de médicaments, sa concentration dans le corps fluctue continuellement. Après avoir reçu une certaine dose, il augmente initialement, puis diminue progressivement, atteignant des valeurs minimales avant la prochaine administration du médicament. Les oscillations de la concentration sont les plus importantes que la plus grande que la dose de médicament et l'intervalle entre les introductions.
Intervalle d'introduction - l'intervalle entre les doses administrées, assurant le maintien de la concentration thérapeutique de la substance dans le sang.
16. Introduction de médicaments à une vitesse constante. La cinétique de la concentration du médicament dans le sang. Concentration stationnaire du sang dans le sang ( CSS.), le temps de sa réalisation, de son calcul et de sa gestion.
 La particularité de l'introduction de HP avec une vitesse constante est un changement en douceur de sa concentration de sang lorsqu'il est administré, tandis que:
La particularité de l'introduction de HP avec une vitesse constante est un changement en douceur de sa concentration de sang lorsqu'il est administré, tandis que:
1) Le temps nécessaire pour atteindre la concentration de médicaments stationnaires est de 4-5T1 et ne dépend pas du taux d'infusion (les valeurs de la dose administrée)
2) Avec une augmentation du taux de perfusion (dose administrée), la valeur de CSS augmente également d'un nombre proportionnel
3) Éliminer les médicaments du corps après la fin de la perfusion occupe 4-5T1.
DESS. - Concentration stationnaire équilibre - la concentration de médicaments atteints au taux d'administration de la vitesse égale de retrait, de sorte:
![]() (de la définition du dédouanement)
(de la définition du dédouanement)
Pour chaque demi-vie ultérieure, la concentration de LS augmente sur la moitié de la concentration restante. Tous ls, obéissant à la loi de l'élimination de la première commande, Va accomplirCSS. Après 4-5 périodes demi-vie.
Approches de la gestion de niveau avecSS.: Changez la dose de LS ou de l'intervalle d'administration
17. Administration de médicaments intermittente. Cinétique de la concentration de la drogue dans le sang, la gamme thérapeutique et toxique de concentrations. Calcul de la concentration fixe ( C.SS.), Frontières de ses oscillations et la gère. Un intervalle adéquat de doses discrètes.
 Fluctuations de la concentration de LS dans le plasma sanguin: 1 - avec une administration gouttière intraveineuse constante; 2 - par une administration fractionnée de la même dose quotidienne à un intervalle de 8 heures; 3 - avec l'introduction d'une dose quotidienne avec un intervalle de 24 heures.
Fluctuations de la concentration de LS dans le plasma sanguin: 1 - avec une administration gouttière intraveineuse constante; 2 - par une administration fractionnée de la même dose quotidienne à un intervalle de 8 heures; 3 - avec l'introduction d'une dose quotidienne avec un intervalle de 24 heures.
Introduction intermittente LS. - Introduction d'une certaine quantité de médicaments à travers des intervalles.
La concentration stationnaire à l'équilibre est réalisée grâce à 4 à 5 périodes de semi-élimination, le temps de sa réalisation ne dépend pas de la dose (au début, lorsque le niveau de concentration de LS est faible, le taux de son élimination est également faible; Comme le montant de sa substance augmente, le taux de son élimination augmente, tellement tôt ou cela arrivera en retard un tel moment où l'augmentation du taux d'élimination équilibre la dose administrée de médicaments et l'augmentation supplémentaire de la concentration pour arrêter)
Le CSS est directement proportionnel à la dose de médicaments et proportionnelle inversement à l'intervalle d'introduction et de dédouanement des médicaments.
Borders oscillation CSS: ![]() ; CSSMIN \u003d CSSMAX × (1 - EM. Fr.). Les fluctuations de la concentration de HP sont proportionnelles à T / T1 / 2.
; CSSMIN \u003d CSSMAX × (1 - EM. Fr.). Les fluctuations de la concentration de HP sont proportionnelles à T / T1 / 2.
Gamme thérapeutique (couloir de sécurité, fenêtre thérapeutique) - Il s'agit d'un intervalle de concentrations de la thérapeutique minimale à l'apparition des premiers signes d'effets secondaires.
Gamme toxique - L'intervalle de concentration du plus haut thérapeutique à la mort.
Mode adéquat d'introduction de doses discrètes: Ce mode d'administration dans lequel la fluctuation de la concentration du médicament dans le sang est placée dans la plage thérapeutique. Pour déterminer le mode d'administration adéquat du médicament, il est nécessaire de calculer. La différence entre CSSmax et CSSMIN ne doit pas nécessairement dépasser 2CSS.
ContrôleCSS.:
Gamme d'oscillationsCSS. Directement proportionnelle à la dose de LS et inversement proportionnelle à l'intervalle de son introduction.
1. Changer la dose de HP: Avec une augmentation de la dose de drogues, la plage d'oscillation que son CSS augmente proportionnellement
2. Changer l'intervalle d'imposition du réseau local: Avec une augmentation de l'intervalle d'administration de la LS, la plage d'oscillation de son CSS est proportionnelle à la diminution
3. Dans le même temps, changez la dose et l'intervalle de l'introduction
18. Dose d'introduction (chargement). Signification thérapeutique, calcul sur les paramètres pharmacocinétiques, les conditions et les restrictions sur son utilisation.
Dose d'introduction (chargement) - Dose administrée en une seule réception et remplissant l'ensemble du volume de la distribution dans la concentration thérapeutique active. VD \u003d (CSS * VD) / F; \u003d mg / l, \u003d l / kg
Signification thérapeutique: La dose d'introduction assure rapidement la concentration thérapeutique active de médicaments dans le sang, ce qui permet, par exemple, d'arrêter rapidement l'attaque, l'arythmie, etc.
La dose d'introduction peut être introduite à la fois uniquement lorsque Ignoré le processus de distribution de la substance
Restreindre l'utilisation du mercredi: Si la distribution de LS se produit Beaucoup plus lentement que son flux sanguinL'introduction de l'ensemble de la dose de démarrage à la fois (surtout par voie intraveineuse) créera une concentration de manière significative au-dessus de la thérapeutique et provoque la survenue d'effets toxiques. La condition d'utiliser le WD: Par conséquent, l'introduction de doses de charge Devrait toujours être lent ou fraction.
19. Soutenir des doses, leur signification thérapeutique et leur calcul pour régime optimal Dosage.
Dose de soutien - Dose LS, introduite systématiquement, qui remplit le volume de dégagement, c'est-à-dire que le fragment VD, qui est effacé de médicaments pour l'intervalle DT: PD \u003d (CSS * CL * DT) / F.
Signification thérapeutique: PD compense la perte avec une autorisation pour l'intervalle entre l'administration du médicament.
Calcul pour une distribution optimale de médicaments (pour une attaque de liaison rapide):
1. Calculez le VD: DR \u003d (CSS * VD) / F
2. Sélectionnez l'intervalle de l'administration de DT (généralement la plupart des LS sont prescrits avec un intervalle proche de T1 / 2) et calculez PD: PD \u003d (CSS * CL * DT) / F
3. Nous vérifions si les oscillations du réseau local dans le sang au-delà de la plage thérapeutique en calculant CSSMAX et CSSMIN: ![]() ; CSSMIN \u003d CSSMAX × (1 - EM. Fr.). La différence entre CSSmax et CSSmin ne doit pas dépasser deux CSS.
; CSSMIN \u003d CSSMAX × (1 - EM. Fr.). La différence entre CSSmax et CSSmin ne doit pas dépasser deux CSS.
La fraction éliminée est à l'horaire (voir B.16) ou par la formule: ![]()
4. Si, avec l'administration du FAI sélectionné, ses oscillations s'étendent au-delà de la gamme thérapeutique, modifient DT et répétent le calcul (paragraphe 2 - paragraphe 4)
NB! Si le réseau local n'est pas destiné à soulager les états urgents ou est accepté dans des comprimés, le VD n'est pas calculé.
20. Différences individuelles, pharmacocinétique des médicaments. Amendements visant à calculer les valeurs individuelles du volume de distribution des médicaments.
1. Différences d'âge Pharmacokenets Médecine.
|
1. La couche cornée de la peau est plus mince, par conséquent, avec l'utilisation anormale des médicaments, le BS est mieux absorbé. L'absorption du réseau local pendant l'utilisation rectale est également meilleure. 2. Le volume de liquide dans le corps des enfants est de 70 à 80%, tandis que les adultes ne sont que de 60%, donc des médicaments hydrophiles VD ont plus de doses plus élevées. 3. Le nouveau-né d'albumine du plasma est inférieur à celui chez les adultes, donc la liaison de médicaments avec des protéines dont ils ont moins intense 4. Dans les nouveau-nés, la faible intensité des systèmes de cytochrome P450 et des enzymes conjuguées, mais la forte activité de systèmes de méthylation. 5. La vitesse de filtration glomérulaire dans les reins reins jusqu'à 6 mois est de 30 à 40% de la vitesse des adultes. L'exercice rénal des médicaments est donc réduit. |
1. Il y a une diminution de la concentration en albumine dans le plasma sanguin et la fraction de médicament associée à la protéine. 2. La teneur en eau dans le corps est réduite de 60% à 45%, par conséquent, le cumul des médicaments lipophiles augmente. 3. La vitesse de filtration glomérulaire peut atteindre 50 à 60% de la vitesse du patient mature, de sorte que l'élimination rénale des médicaments est considérablement limitée. |
2. Sexualité dans l'action des drogues. Pour les femmes, un poids corporel plus petit est caractérisé par rapport aux hommes. Par conséquent, la valeur des doses de médicaments pour eux devrait être, en règle générale, à la limite inférieure de la plage de dose thérapeutique.
3. Etats pathologiques du corps et l'effet des drogues
A) Maladie du foie: F médicaments en raison de la fermeture du métabolisme prêté, une fraction de LS non liée en raison d'un manque de synthèse des albumines, les effets des médicaments sont prolongés en raison de leur biotransformation.
B) pathologie des reins: ralentit l'élimination des médicaments, qui sont dérivés à travers les reins
4. Facteurs génétiques - une pénurie de celles ou d'autres enzymes de métabolisme LS peuvent contribuer à la prolongation de leur action (pseudocholinestérase, etc.)
Amendements visant à calculer les valeurs individuelles du volume de distribution des médicaments:
A) Lorsque les médicaments lipophobes d'obésité ne sont pas solubles dans le tissu adipeux þ doivent être calculés poids parfait Par croissance (formule Brock: le poids idéal \u003d croissance (en cm) - 100) et recalculer VD pour le poids idéal de la croissance.
B) à l'œdème, il est nécessaire de calculer l'excès de volume d'eau \u003d en surpoids - Parfait, VD devrait être augmenté par un litre de chaque kilogramme excessif d'eau.
La dépendance des principaux paramètres pharmacocinétiques de divers facteurs:
1. Aspiration LS: À l'âge de l'absorption des médicaments, son métabolisme au cours de la pré-élimination, la biodisponibilité des médicaments change.
2. Volume de la distribution VD: ¯ avec l'âge et l'obésité, lors de gonflement
3. La demi-vie: varie avec l'âge et lorsque l'obésité (puisque VD diminue)
4. Dégagement: déterminé par l'état fonctionnel des reins et du foie
21. Dégagement rénal des médicaments, des mécanismes, de leurs caractéristiques quantitatives et qualitatives.
La clairance du rein est une mesure du volume plasmatique sanguin, qui est purifié à partir de la substance médicinale par unité de temps par les reins: cl (ml / min) \u003d u × v / p, où vous êtes la concentration de ls dans ml de L'urine, V est le volume d'urine attribué dans les mines et la concentration de P \u003d LS en ml de plasma.
Mécanismes de dédouanement rénal et de leurs caractéristiques:
1. Filtration: LS, alloué Seulement filtrage (L'insuline) aura une clairance égale à SCF (125-130 ml / min)
Déterminé par: flux sanguin rénal, fraction ls non liée et capacité de filtration du rein.
La plupart des médicaments ont des poids moléculaires bas et donc librement filtrés du plasma dans le glomérulaire.
2. Sécrétion active: LS, alloué Filtration et sécrétion complète (l'acide para-aminohypruury) aura une clairance égale à la clairance plasmatique rénale (650 ml / min)
Le canal de couchage contient Deux systèmes de transportqui peut mettre en évidence la drogue dans ultrafiltrate, une pour Acides organiques Et l'autre pour Terrain bio. Ces systèmes nécessitent de l'énergie pour le transport actif contre le gradient de concentration; Ils sont le lieu de la concurrence pour le transporteur de certaines substances médicinales avec d'autres.
Déterminé: vitesse de sécrétion maximale, urine
3. Réabsorption: Les valeurs de la clairance entre 130 et 650 ml / min suggèrent que ls Filtré, démarquent et partiellement reconstruit
La réabsorption se produit tout au long du canal rénal et dépend de la polarité du médicament, non polaire et lipophile recule.
Déterminé: la taille du pH primaire et ionisation des médicaments
Un certain nombre d'indicateurs tels que Âge, utilisation conjointe de plusieurs médicaments, maladie affecter de manière significative la clairance des reins:
A) Échec rénal ® LS® Réduction de la clairance haut niveau Ls dans le sang
B) la perte de protéines sériques glomérulonéphrite ®, généralement disponible et connectée LS ® augmente le niveau de fraction LS gratuite dans le plasma
22. Facteurs affectant le dédouanement des drogues du rein. La dépendance de la clairance des propriétés physicochimiques des médicaments.
Facteurs affectant la rénaleCl.:
A) filtrage glomérulaire
B) débit sanguin rénal
C) vitesse maximale de la sécrétion
D) urine
E) fraction non liée dans le sang
Dépendance de la clairance rénale des propriétés physicochimiques de la LV:
Modèles généraux:1) Les LS Polar ne sont pas facinables, non polaires - 2) Ion LS Secrétée, pas ionique - non sécrétée.
I. Substances non polaires non ioniques: filtré uniquement sous des formes non liées, n'est pas sécrétée, réabsorbée
Le dédouanement est petit et déterminé: a) la fraction de LS, sans rapport dans le sang B) urine
II. Substances polaires non ioniques: filtré sous forme non liée, non sécrétée, ne réabsorbe pas
La clairance du rein est élevée, déterminée par: a) la fraction LS non liée dans le sang b) la vitesse du filtrage glomérulaire
III. Ionisé dans l'urine ne sont pas polaires sous forme non ionique: filtrée, activement sécrétée, non polaire réabsorbée
Le dédouanement du rein est déterminé par: a) la fraction LS, sans rapport dans le sang b) la fraction de LS, ionisée dans l'urine dans le volume d'urine
Iv. Urine polaire ionisée sous forme non ionisée: filtrée, active activement, ne réabsorbe pas
Le dédouanement est déterminé: a) Flux sanguin rénal et vitesse de filtrage glomérulaire B) Vitesse maximale de sécrétion
23. Dégagement du foie des médicaments, ses déterminants et ses restrictions. Cycle de drogue entérogènes.
Mécanismes de dégagement hépatiques:
1) Métabolisme (biotransformation) par oxydation, récupération, alkylation, hydrolyse, conjugaison, etc.
La stratégie principale de métabolisme xénobiotique: métabolites polaires non polaires (hydrophiles) (hydrophile) affichés avec urine.
2) Sécrétion (élimination des substances non tratancées en bile)
Seules des substances polaires à poids moléculaire\u003e 250 actifs sont transportées en bile (acides organiques, bases).
Déterminants du dédouanement hépatique:
A) la vitesse du flux sanguin dans le foie
B) Vitesse maximum excrétion ou transformations métaboliques
C) km - mikhailis constante
D) sans rapport avec la fraction de protéines
Limitations de l'autorisation du foie:
1. Si vmax / km est VELIKO → CL ... saignement de la vitesse dans le foie
2. Si Vmax / km moyennement → CL \u003d somme de tous les facteurs
3. Si vmax / km Petit → Le four CL est petit, limité
CYCLE INTÉGATISEPATIQUE LS -Un certain nombre de médicaments et de produits de leur transformation de manière significative sont excrétés dans une biliaire dans l'intestin, d'où il est partiellement excrété d'excréments, et en partie - Re-absorbé dans le sang, réitère au foie et est affiché dans l'intestin.
L'élimination hépatique des médicaments peut être considérablement modifiée Maladie du foie, âge, régimes, génétique, durée de la drogue (Par exemple, en raison de l'induction d'enzymes hépatiques) et d'autres facteurs.
24. Facteurs changeant la clairance des substances médicinales.
1. Interactions de LS au niveau: sécrétion rénale, transformation biochimique, phénomènes d'induction enzymatiques
2. Maladies des reins: troubles du flux sanguin, lésions rénales aiguës et chroniques, résultats de maladies rénales longues
3. Maladies du foie: cirrhose alcoolique, cirrhose primaire, hépatite, hépatomes
4. Maladies des organes du GCT et des organes endocriniens
5. Intolérance individuelle (pas d'enzymes d'acétylation - intolérance à l'aspirine)
25. Correction de la thérapie médicinale pour les maladies du foie et des reins. Approches générales. Correction du mode de dosage sous le contrôle de la clairance générale du médicament.
1. Annuler les médicaments qui ne sont pas nécessaires
2. Avec des maladies rénales, utilisez des médicaments qui se distinguent du foie et inversement.
3. Réduisez la dose ou augmentez l'intervalle entre les introductions
4. Surveillance minutieuse pour les effets secondaires et toxiques
5. En l'absence d'effet pharmacologique, soulevez la dose, il est nécessaire de contrôler lentement et sous le contrôle des effets pharmacologiques et toxiques.
6. Si possible, déterminez la concentration de la substance dans le plasma et corréler la thérapie pour le CL du médicament individuellement
7. Utilisez un moyen indirect pour évaluer cl.
Correction du mode de dosage sous le contrôle de la clairance globale du médicament:
Correction de dose : Dind. \u003d DutyN. × Clind / Clutpichn.
Avec l'administration de médicaments intraveineux continue: Introduction Speed \u200b\u200bIndividuel \u003d Introduction Vitesse typique × Cl PRI. / Cl est typique.
Lorsque l'introduction intermittente: 1) Modifiez la dose 2) Modifier l'intervalle 3) Modifier les deux paramètres. Par exemple, avec une diminution de la clairance de 50%, il peut être réduit de 50% de la dose et économisez l'intervalle de 50% ou d'augmenter l'intervalle deux fois et de sauvegarder la dose. Il est préférable de réduire la dose et de conserver l'intervalle d'administration.
26. Correction du mode de dosage sous le contrôle de la fonction rénale résiduelle.
Clément Creinina - l'indicateur quantitatif le plus important de la fonction rénale, sur la base desquels vous pouvez affirmer le mode de dosage
Nous savons:
A) Fonction rénale résiduelle définissant la clairance de la créatinine chez ce patient CLC / patient
B) clairance générale de ce médicament (CLLS / Généralités) et la quote-part du dédouanement des médicaments en général
C) clairance de créatinine normale Clkr / Norm par Normogramme
3) CSS et F pour ce réseau local (dans le répertoire)
Trouver: Dose de ls pour ce patient
CLLS / Taux rénal \u003d CLLS / Total X Part du dédouanement de rein LS en clairance générale
CLLS / Patient rénal \u003d CLKR / Patient / SLKR / NORME * CLLS / Taux rénal
CLLS / NON RENISE NORME \u003d CLLS / TOTAL - CLLS / Taux rénal
CLLS / TOTAL PATIENT \u003d CLLS / PATIENT RENAL + CLLS / NO TAUX RENAL
La dose de ce ls à l'intérieur avec une fonction de rein normale est égale à: pdnmm \u003d CSS x cl / f
La dose de ce ls à l'intérieur de notre patient est égale: pdbology \u003d pdnmm x CLLS / Patient commun / CLLS / Général
Répondre: Padbolna
27. Correction du traitement médicamenteux dans les dommages causés au foie et à d'autres conditions pathologiques.
Les maladies du foie peuvent réduire la clairance et allonger la demi-vie de nombreux LS. Cependant, certains médicaments éliminés par le foie ne modifient pas ces indicateurs lorsqu'ils sont dotés de la fonction de foie altérée, donc Les maladies du foie n'affectent pas toujours leur propre dédouanement du foie. Actuellement, il n'existe pas de marqueur fiable qui pourrait être utilisé pour prédire le dédouanement hépatique de cette clairance de la créatinine.
Correction du régime de dosage de la maladie rénale, voir ci-dessus en B.26, principes généraux de la correction - B.25.
28. Stratégie de traitement médicamenteuse individuelle.
La reconnaissance d'un rôle important de la concentration en tant que liant de pharmacocinétique et de pharmacodynamique contribue à la création d'une stratégie de concentration cible - optimisation de la dose dans un patient donné basé sur la mesure de la concentration de LS. Il se compose des étapes suivantes:
1. Sélection de la concentration de la cible
2. Calcul de VD et CL basé sur des valeurs typiques et des modifications afin de prendre en compte de tels facteurs que le poids corporel et la fonction rénale.
3. Entrer dans la dose de démarrage ou la dose de maintenance, calculée sur la base des valeurs de TC, VD et CL.
4. Enregistrement de la réaction du patient et la définition de la concentration de LS
5. Révision VD et CL basée sur les résultats de la mesure de la concentration.
6. Répétez les étapes 3-6 dans le but de choisir la dose nécessaire requise pour la réaction optimale.
29. Biotransformation des médicaments, son sens biologique, l'orientation principale et l'effet sur l'activité des médicaments. Les principales phases des transformations métaboliques de médicaments dans le corps.
Biotransformation de LS. - Transformations chimiques de LS dans le corps.
Signification biologique de la biotransformation LS: Créer un substrat pratique pour une élimination ultérieure (en tant que matériau énergétique ou plastique) ou dans l'accélération de l'élimination de la LS du corps.
L'objectif principal des transformations métaboliques de la LS: LS non polaire → Les métabolites polaires (hydrophiles) dérivés d'urine.
Les deux phases des réactions métaboliques des médicaments sont distinguées:
1) Transformation métabolique (Réactions non sécrétiques, phase 1) - Transformations de substances dues à l'oxydation microsomale et éminomique, la récupération et l'hydrolyse
2) Conjugaison (réactions synthétiques, phase 2) - processus biosynthétique, accompagné d'un ajout au médicament ou de ses métabolites d'un certain nombre de groupes chimiques ou de molécules de composés endogènes par a) de la formation de glucuronides b) des esters de glycérol) sulfoockets d) acétylation d) méthylation
L'influence de la biotransformation sur l'activité pharmacologique de la LAN:
1) Le plus souvent, les métabolites de la biotransformation ne possèdent pas une activité pharmacologique ou leur activité est réduite par rapport à la matière de départ
2) Dans certains cas, les métabolites peuvent maintenir une activité et même dépasser l'activité de la substance source (la codéine est métabolisée à une morphine plus active pharmacologique)
3) Parfois, des substances toxiques sont formées pendant la biotransformation (métabolites d'isoiazide, lidocaïne)
4) Parfois, des métabolites avec des propriétés pharmacologiques opposées sont formées lors de la biotransformation (métabolites de b2-adrénorécepteurs b2 non sélectifs possèdent les propriétés de ces bloqueurs de récepteurs)
5) Un certain nombre de substances sont des prodrugs qui ne donnent initialement pas d'effets pharmacologiques, mais pendant la biotransformation est convertie au BAV (inactif L-Dope, pénétré par la BC, se transforme en un cerveau en dopamine active, tandis qu'il n'y a pas d'effets systémiques de dopamine).
30. La signification clinique de la biotransformation des médicaments. Influence du sol, âge, poids corporel, facteurs environnementaux, fumeur, alcool sur la biotransformation des médicaments.
Valeur clinique de la biotransformation des médicaments: Etc. La dose et la fréquence de réception nécessaire pour obtenir une concentration efficace dans le sang et les tissus peuvent varier chez les patients en raison des différences individuelles de la distribution, du taux métabolique et de l'élimination de la LS, sont importantes dans la pratique clinique.
Impact sur la biotransformation de LS de divers facteurs:
MAIS) État fonctionnel foie: Avec ses maladies, la clairance LS est généralement réduite et la période de semi-élimination augmente.
B) Effet des facteurs environnementaux: Le tabagisme contribue à l'induction du cytochrome P450, à la suite de laquelle le métabolisme des médicaments est accéléré lors de l'oxydation microsomale
DANS) Aux végétariens Biotransformation LAN a ralenti
D) les personnes âgées et les jeunes patients sont caractéristiques sensibilité accrue À l'effet pharmacologique ou toxique des médicaments (chez les personnes âgées et les enfants de moins de 6 mois, l'activité de l'oxydation microsomale est réduite)
E) Le métabolisme des hommes de certains HP se produit plus rapidement que les femmes, car les androgènes stimulent la synthèse d'enzymes de foie microsomal (éthanol)
E) Teneur élevée dans les protéines alimentaires et intense stress de l'exercice : Accélération du métabolisme des médicaments.
G) Alcool et obésité ralentir le métabolisme de la LS.
31. Interaction métabolique des médicaments. Maladies affectant leur biotransformation.
Interaction métabolique des médicaments:
1) L'induction des enzymes de métabolisme de la fusion est une augmentation absolue de leur quantité et de leur activité en raison de l'impact de certains LS. L'induction conduit à l'accélération du métabolisme de LS et (en règle générale, mais pas toujours) à une diminution de leur activité pharmacologique (rifampicine, barbituriques - inducteurs de cytochrome P450)
2) Inhibition des enzymes du métabolisme de la LS - L'oppression de l'activité des enzymes de métabolisme sous l'action de certains xénobiotiques:
A) Interaction métabolique compétitive - HP avec une grande affinité à certaines enzymes réduisent le métabolisme de LS avec une affinité plus faible de ces enzymes (Verapamil)
B) Reliaison à un génome induisant la synthèse de certains isoenzyme cytochrome p450 (TSIMIDEN)
C) Inactivation directe des isoenzymes du cytochrome P450 (flavonoïdes)
Maladies affectant le métabolisme de la LAN:
A) maladie rénale (perturbation du flux sanguin rénal, net et maladies chroniques Reins, résultats des longues maladies rénales)
B) Maladie du foie (cirrhose primaire et alcoolique, hépatite, hépatome)
C) Maladies des organes gastro-intestinaux et endocriniens
C) Intolérance individuelle de certains LS (pas d'enzymes d'acétylation - intolérance à l'aspirine)
32. Chemins et mécanismes d'élimination des médicaments du corps. Possibilités de gérer la drogue.
Chemins et mécanismes d'élimination des médicaments: Élimination du foie et des reins de la LAN et d'autres organes:
A) reins par filtration, sécrétion, réabsorption
B) foie par biotransformation, excrétion avec bile
C) à travers les poumons, la salive, la sueur, le lait, etc. par sécrétion, évaporation
Les possibilités de gérer les processus de suppression des médicaments:
1. Contrôle du pH: Dans l'urine alcaline, l'élimination des composés acides augmente, en élimination acide des composés principaux
2. Application de médicaments cholérétiques (Holenzim, Allohol)
3. Hémodialyse, dialyse péritonéale, hémosorption, lymphosorption
4. Dirésis forcés (dans / dans NaCl ou glucose pour la charge d'eau + furosémide ou mannitol)
5. Laver l'estomac, appliquer un lavement
33. Concept de récepteurs en pharmacologie, nature moléculaire des récepteurs, mécanismes de signalisation des médicaments (types de signalisation transmembranaire et intermédiaires secondaires).
Récepteurs -Composants moléculaires d'une cellule ou d'un organisme qui interagissent avec des médicaments et induisent un certain nombre d'événements biochimiques menant au développement de l'effet pharmacologique.
Le concept de récepteurs en pharmacologie:
1. Les récepteurs déterminent les modèles quantitatifs de l'action LS
2. Les récepteurs sont responsables de la sélectivité de l'action LS
3. Intermédiaires des récepteurs d'antagonistes pharmacologiques
Le concept de récepteurs - la base de l'utilisation initiale des médicaments affectant les processus et les communications réglementaires, biochimiques.
Nature du récepteur moléculaire:
1. Protéines réglementaires, médiateurs d'action de divers signaux chimiques: neurotransmetteurs, hormones, autocoïdes
2. Porte-protéines enzymes et protéines transmembranaires (Na +, K + ATPAZ)
3. Protéines structurelles (tubuline, protéines cytosquelette, surface cellulaire)
4. Protéines nucléaires et acides nucléiques
Mécanismes de signalisation d'action antidrogue:
1) Pénétration de ligand soluble dans les lipides à travers la membrane et leur effet sur les récepteurs intracellulaires.
2) La molécule de signal se lie au domaine extracellulaire de la protéine transmembranaire et active l'activité enzymatique de son domaine cytoplasmique.
3) La molécule de signal est associée au canal d'ions et ajuste son ouverture.
4) La molécule de signal se lie au récepteur de la surface de la cellule, qui est conjugué avec l'enzyme effecteur par la protéine G. G-protéine active le médiateur secondaire.
Types d'alarme transmembranaire:
A) à travers des récepteurs de 1 TMS possédant et ne possédant pas une activité de tyrosine kinase
B) à travers des récepteurs 7-TMS associés à la protéine G
C) à travers des canaux d'ions (contacts fendus à charge potentiels, dépendants du ligand)
Intermédiaires secondaires: Tsamf, ions CA2 +, Dag, if3.
34. Mécanismes physico-chimiques et chimiques de drogues.
MAIS) Interaction physico-chimique avec biosubstrate - action néelectrique.
Les principaux effets pharmacologiques: 1) Narcotic 2) Dépressives générale 3) Paralysing 4) Action de membronolytique 5).
Substances naturelles chimiques: hydrocarbures chimiquement inertes, esters, alcools, aldéhydes, barbituriques, narcotique de gaz
Le mécanisme d'action est une membranes de dzing réversible.
B) Chimique Mécanisme (biochimique moléculaire) d'action de drogue.
Les principaux types d'interaction chimique avec biosubstrate:
- Interactions faibles (non-repas, réversibles) (hydrogène, ionique, monodipole, hydrophobe).
- Liaisons covalentes (alkylation).
La signification des interactions non covalentes du réseau local: L'action est non spécifiée, ne dépend pas de la structure chimique de la substance.
La valeur des interactions covalentes des médicaments: action spécifiquement, dépend de manière critique de bâtiments chimiquesest mis en œuvre par influence sur les récepteurs.
35. Termes et concepts de pharmacologie quantitative: effet, efficacité, activité, agoniste (complet, partiel), antagoniste. Différence clinique des concepts Activité et efficacité des médicaments.
 Effet (réponse)- le rendement quantitatif de la réaction de l'interaction de la cellule, de l'organe, du système ou du corps avec un agent pharmacologique.
Effet (réponse)- le rendement quantitatif de la réaction de l'interaction de la cellule, de l'organe, du système ou du corps avec un agent pharmacologique.
Efficacité - mesure de la réaction le long de l'axe d'effet - la valeur de la réponse du système biologique à l'effet pharmacologique; C'est la capacité des médicaments de rendre l'action maximale possible pour cela.. En fait, c'est l'effet maximal de l'effet pouvant être obtenu lorsque ce médicament est introduit. Normalement caractérisé par la valeur d'Emax. L'EMAX plus élevé, plus l'efficacité du médicament est élevée.
Activité - La mesure de la sensibilité aux médicaments le long de l'axe de concentration caractérise l'affinité (affinité du ligand au récepteur) montre comment la dose (la concentration) du médicament est capable de développer un effet standard égal à 50% du médicament maximal possible. Normalement caractérisé par EC50 ou ED50. Plus l'activité de médicaments est élevée, plus la dose est réduite pour reproduire l'effet thérapeutique.
Efficacité: 1 \u003d 2\u003e 3
Activité: 1\u003e 3\u003e 2
Dans les activités cliniques, il est plus important de connaître l'efficacité et non une activité, car nous sommes plus intéressés par la capacité du réseau local à causer un certain effet dans le corps.
Agoniste- Ligand, qui lie au récepteur et provoque une réponse biologique, déclenchant le système physiologique. Agoniste complet- réponse maximale, Partiel - provoquer une réaction plus petite même lorsque tous les récepteurs occupent.
Antagoniste - Les ligands occupent des récepteurs ou les modifient de manière à perdre la capacité d'interagir avec d'autres ligands, mais ne provoquent pas de réaction biologique (action block d'agonistes).
 Antagonistes compétitifs- Interagissez avec les récepteurs de manière réversible et concurrentiellement des agonistes. Une augmentation de la concentration d'agoniste peut complètement éliminer l'effet antagoniste. L'antagoniste concurrentiel déplace la courbe "dose-effet" pour l'agoniste, augmente l'EC50 n'affecte pas EMAX.
Antagonistes compétitifs- Interagissez avec les récepteurs de manière réversible et concurrentiellement des agonistes. Une augmentation de la concentration d'agoniste peut complètement éliminer l'effet antagoniste. L'antagoniste concurrentiel déplace la courbe "dose-effet" pour l'agoniste, augmente l'EC50 n'affecte pas EMAX.
Antagonistes non concurrentiels- modifier de manière irréversible l'affinité des récepteurs à l'agoniste, la liaison ne se produit souvent pas avec le secteur actif du récepteur, une augmentation de la concentration de l'agoniste n'élimine pas l'action de l'antagoniste. L'antagoniste non concurrentiel réduit EMAX, ne change pas le CE50, la courbe "dose-effet" est compressée par rapport à l'axe vertical.
36. Lois quantitatives de l'action des médicaments. Loi de réduire la réponse des systèmes biologiques. Modèle Clark et sa conséquence. Le type général de concentration de dépendance est l'effet dans les coordonnées normales et lognormales.
Clark-Ariens Modèle:
1. L'interaction entre le ligand (L) et le récepteur (R) est réversible.
2. Tous les récepteurs de ce ligand sont équivalents et indépendants (leur saturation n'affecte pas les autres récepteurs).
3. L'effet est directement proportionnel au nombre de récepteurs occupés.
4. Le ligand existe dans deux états: libre et associé au récepteur.
MAIS) ![]() Lorsque KD est une constante d'équilibre, une activité interne KE.
Lorsque KD est une constante d'équilibre, une activité interne KE.
B) T. K. À titre d'augmentation du nombre de ligands à un moment donné, tous les récepteurs seront occupés, puis le nombre maximum possible de complexes récepteurs de ligand éduqués est décrit par la formule:
\u003d [R] × ![]() (1)
(1)
L'effet est déterminé par la probabilité d'activation du récepteur lors de la liaison au ligand, c'est-à-dire son activité interne (KE), donc e \u003d ke ×. Dans ce cas, l'effet est maximum à clé \u003d 1 et minimal et ke \u003d 0. Naturellement, l'effet maximum est décrit par le rapport EMAX \u003d KE ×, où - le nombre total de récepteurs pour ce ligand
L'effet dépend de la concentration de ligand sur les récepteurs [S], donc
E \u003d empax ![]() (2)
(2)
Des relations ci-dessus, il suit que EC50 \u003d KD
![]()
EMAX est l'effet maximum, BMAX est le nombre maximum de récepteurs connexes, EC50 est la concentration de la LS, dans laquelle l'effet se produit égal à la moitié du maximum, KD est la constante de dissociation de la substance du récepteur auquel 50% des récepteurs sont associés.
La loi de la diminution de la réponse Conforme à la dépendance parabolique «Concentration - efficacité». La réponse aux petites doses de drogue augmente généralement directement proportionnelle à la dose. Toutefois, avec une augmentation de la dose, la croissance de la réponse est réduite et finalement une dose peut être obtenue, à laquelle il n'ya plus d'augmentation de la réponse (en raison de l'occupation de tous les récepteurs de ce ligand).
37. Changer l'effet des médicaments. Effet d'effets gradués et quantiques, Essence et applications cliniques. Les mesures Évaluation quantitative L'activité et l'efficacité des médicaments dans la pratique expérimentale et clinique.
Tous les effets pharmacologiques peuvent être divisés en deux catégories:
MAIS) Effets graduels (continus, intégrales) - de tels effets des médicaments pouvant être mesurés quantitativement (l'effet des médicaments anti-hypotenseurs - au niveau de la pression artérielle). Décrit la "courbe de dose-effet" progressive (voir. 36), sur la base desquelles il est possible d'estimer: 1) Sensibilité individuelle à LS 2) Activité LAN 3) Efficacité maximale des médicaments
B) Effets quantiques - Ces effets des médicaments, qui sont une valeur discrète, une caractéristique qualitative, sont décrites uniquement par plusieurs options pour les états (mal de tête après avoir reçu des analgésiques ou y est ou non). La courbe quantique de la dose-effet est décrite, où la dépendance de l'effet dans la population de la valeur de la dose reçue de LS est décrite. Le graphique de la dépendance à effet de dose a une forme en forme de dôme et est identique à la courbe gaussienne de la distribution normale. Sur la base de la courbe quantique, il est possible: 1) Évaluer la sensibilité de la population de LS 2) Notez la présence de l'effet à cette dose 3) Sélectionnez la dose thérapeutique moyenne.
Différences entre la caractéristique progressive et quantique de "dose-effet":
Une évaluation quantitative de l'activité et de l'efficacité des médicaments est effectuée sur la base de la construction des courbes "effet de dose" et de leur évaluation ultérieure (voir B.35)
38. Types d'action des médicaments. Changer l'action des médicaments lorsqu'ils sont ré-administrés.
Types d'action LAN:
1. Action locale - action d'une substance découlant sur la place de son application (anesthésique - sur la membrane muqueuse)
2. Action de résorbation (système) - L'effet de la substance se développe après son aspiration, son admission au flux sanguin global, puis dans le tissu. Cela dépend des itinéraires d'injection de médicaments et de leur capacité à pénétrer des barrières biologiques.
À la fois aux médicaments d'action locale et résorbative peuvent fournir soit DirectSoit Réflexe Influence:
A) Influence directe - Contact direct avec l'organe cible (adrénaline sur le cœur).
B) Reflex - Modification de la fonction d'organes ou de centres nerveux en influençant les extérieurs et les interorécteurs (morceaux de moutarde dans la pathologie des organes respiratoires améliorent réflexivement leur trophique)
Changements dans l'action des médicaments lors de leur ré-administration:
1. Cumul- une augmentation de l'effet due à l'accumulation dans le corps du réseau local:
a) Cumulations matérielles - Accumulation de substance active dans le corps (glycosides coeur)
b) Cumulement fonctionnel - Augmentation des changements dans la fonction des systèmes corporels (modification de la fonction du CNS dans l'alcoolisme chronique).
2. Tolérance (dépendance) -Réduire la réponse de l'organisme sur l'introduction répétée de médicaments; Afin de restaurer la réaction au réseau local, il doit être introduit dans toutes les doses de plus en plus (diazépam):
A) Véritable tolérance - observée à la fois avec l'administration entérale et dans l'administration parentérale des médicaments, ne dépend pas du degré d'aspiration dans le flux sanguin. Il est basé sur des mécanismes pharmacodynamiques de la dépendance:
1) désensibilisation - une diminution de la sensibilité du récepteur au médicament (B-Adrreminimetics à une utilisation à long terme conduit à la phosphorylation des adrénorécepteurs b-adrénorécepteurs qui ne sont pas capables de réagir à la b-adreminimétique)
2) La réglementation DOWN est une diminution du nombre de récepteurs au médicament (avec une administration répétée d'analgésiques narcotiques, le nombre de récepteurs opioïdes diminue et de plus en plus de doses de médicament sont nécessaires pour provoquer la réponse souhaitée). Si la drogue bloque les récepteurs, le mécanisme de tolérance à celui-ci peut être associé à une régulation en hausse - une augmentation du nombre de récepteurs au médicament (B-adrénoblocators)
3) L'inclusion de mécanismes de régulation compensatoire (avec des administrations répétées de médicaments anti-hypotensissants, effondrement se produit de manière significative moins fréquente qu'à la première introduction en raison de l'adaptation des barorécepteurs)
B) la tolérance relative (pseudoolence) - ne se développe que lorsque la LS est introduite à l'intérieur et est associée à une diminution de la vitesse et de l'exhaustivité de l'aspiration du médicament
3. Tahofilaxie - une condition dans laquelle l'administration fréquente de médicaments provoque le développement de la tolérance dans quelques heures, mais à des injections suffisamment rares de la LC, son effet est entièrement entretenu. Le développement de la tolérance est généralement associé à l'épuisement des systèmes effectrices.
4. La toxicomanie - Un désir irrésistible pour la réception de la substance introduite plus tôt. Les dépendances mentales (de la cocaïne) et physiques (morphine) sont isolées.
5. Hypersensibilité - une réponse allergique ou une autre réponse immunologique aux médicaments lors de la ré-administration.
39. La dépendance de l'action des médicaments d'âge, de genre et caractéristiques individuelles organisme. La valeur des rythmes quotidiens.
MAIS) Par âge: chez les enfants et dans la sensibilité des personnes âgées à la drogue augmenté (car les enfants, il y a une insuffisance de nombreuses enzymes, des fonctions rénales, une hausse perméabilité de la Colombie-Britannique, chez les personnes âgées, l'absorption des médicaments est ralentissement, le métabolisme est moins efficace, le métabolisme est abaissé par des drogues rénales):
|
1. Les nouveau-nés ont réduit la sensibilité aux glycosides cardiaques, car elles sont par unité de zone cardiomyocytaire supérieure à Na + / k + -atfaz (cibles de glycosides). 2. Les enfants sont inférieurs à la sensibilité à la succinylcholine et à l'atrakurie, mais la sensibilité à tous les autres relaxants musculaires est augmenté. 3. Les agents psychotropes peuvent provoquer des réactions anormales chez les enfants: psychostimulants - peut augmenter la concentration d'attention et réduire l'hyperactivité du moteur, les tranquillisants - au contraire, sont en mesure de provoquer la t. N. N. Excitation atypique. |
1. La sensibilité aux glycosides cardiaques augmente fortement grâce à une diminution du numéro NA + / K + -ATFAZ. 2. La sensibilité aux b-adrénoblocers est réduite. 3. Augmente la sensibilité aux bloqueurs de canaux de calcium, car Barraflex est affaibli. 4. Une réaction atypique aux médicaments psychotropes, une telle réaction est notée. |
B) De Paul:
1) des moyens hypotensens - clonidine, b-adrénoblyses, diurétiques peuvent causer des violations fonctions sexuelles Chez les hommes, mais n'affecte pas le travail système reproducteur Femmes.
2) Les stéroïdes anabolisants entraînent un effet plus important dans le corps des femmes que dans le corps des hommes.
DANS) Des caractéristiques individuelles du corps: La carence ou l'excès de certaines enzymes de métabolisme LS entraîne une augmentation ou une diminution de leur action (carence en pseudo-oscillatère de sang - anomaliment à long terme morlaxation avec succinylcholine)
RÉ) Des rythmes quotidiens: Changer l'action des médicaments sur le corps quantitativement et qualitativement en fonction de l'heure de la journée (action maximale à l'activité maximale).
40. Variabilité et variabilité de l'action du médicament. Hypo et hyperréactivité, tolérance et tachofilaxie, hypersensibilité et idiosyncrasie. Causes de la variabilité des médicaments et de la stratégie de thérapie rationnelle.
Variabilité Reflète la différence entre les individus en réponse à ce médicament.
Causes de la variabilité de l'action LS:
1) Changement de la concentration de la substance dans la zone de récepteur - en raison des différences de taux d'aspiration, de sa distribution, de son métabolisme, de l'élimination
2) Variations à la concentration du ligand endogène du récepteur - Propranolol (β-adrénoblocilité) ralentit la fréquence cardiaque chez les personnes présentant des taux élevés de catécholamines dans le sang, mais n'affecte pas la fréquence cardiaque de fond aux athlètes.
3) Modifiez la fonction de densité ou de récepteur.
4) Modification des composants de la réaction située distale que le récepteur.
Thérapie de stratégie rationnelle: Objectif et posologie de drogues, en tenant compte des causes ci-dessus de la variabilité de l'action LS.
Hyporactivité - réduire l'effet de cette dose de médicaments par rapport à l'effet observé chez la plupart des patients. Hyperactivité - Améliorer l'effet de cette dose de médicaments par rapport à l'effet observé chez la plupart des patients.
Tolérance, tachofilaxie, hypersensibilité - voir B.38
Particularité - Réponse pervers de l'organisme sur ce médicament, associée aux caractéristiques génétiques des métabolismes de LS ou de réactivité immunologique individuelle, y compris les réactions allergiques.
41. Évaluation de la sécurité des médicaments. Index thérapeutique et limites de sécurité standard.
L'évaluation de la sécurité est effectuée sur deux niveaux:
A) préclinique (obtenant des informations sur la toxicité des médicaments, influence sur fonctions de reproduction, Embryotoxicité et tératogénicité, effets distants)
B) Clinique (évaluation ultérieure de l'efficacité et de la sécurité des médicaments)
Si après l'effet du plateau est atteint, la dose de LS continuera à croître, puis à une certaine période, il commencera à manifester l'effet toxique. La dépendance de l'effet toxique sur la dose (concentration) de la LS est du même caractère que son effet utile et peut être décrit par des courbes progressives ou quantiques. Sur ces courbes, la valeur peut également être déterminée. Td.50 ou tc50 - Dose toxique (concentration) de médicaments, qui provoque un effet toxique égal à 50% du maximum (pour une courbe quantique - effet toxique sur 50% des personnes de la population). Parfois, au lieu de TD50, utilisez l'indicateur Ld.50 - dose de volqui provoque la mort de 50% des objets de la population.
L'évaluation de la sécurité de la LS est caractérisée sur la base des courbes progressives ou quantiques de "effet de dose" et des indicateurs suivants:
MAIS) Index thérapeutique - Il s'agit du rapport entre les doses toxiques et efficaces de médicaments, qui provoquent l'apparition d'un effet semi-maximal: TI \u003d TD50 / ED50. Plus l'ampleur de l'indice thérapeutique est grande, plus le médicament est sécurisé.
B) Latitude thérapeutique (fenêtre thérapeutique) - Il s'agit d'une gamme de doses entre les doses minimales thérapeutiques et minimales toxiques de médicaments. Il s'agit d'un indicateur de sécurité plus correct des médicaments, car il permet de prendre en compte le degré croissant d'effets indésirables sur la courbe "effet de dose".
DANS) Facteur de sécurité fiable - Il s'agit du rapport de la dose toxique minimale au maximum efficace (FNB \u003d TD1 / ED99), montre combien de fois la dose thérapeutique de médicaments sans risque de développement d'une indexation (effets indésirables) peut être dépassée.
RÉ) Couloir thérapeutique - Il s'agit de la gamme de concentrations de médicaments efficaces dans le sang, qui doivent être créées et maintenues dans le corps afin d'atteindre l'action thérapeutique souhaitée.
42.46. L'interaction des médicaments. Incompatibilité des médicaments (car les questions sont interdépendantes, choisissez par circonstances)
Interaction LS - Il s'agit d'un changement de gravité et de la nature des effets tout en utilisant simultanément ou par l'utilisation préliminaire de plusieurs médicaments.
Causes des interactions indésirables:
1) Polypragmasia - 6 Et plus LS donne des effets secondaires de 7 fois plus que si la LS est inférieure à 6.
2) erreurs de médecins
3) violation du mode dosage
Justification de la thérapie combinée:
1. La monothérapie n'est pas assez efficace.
2. L'absence avec la plupart des maladies de la thérapie étiotropique þ Le besoin d'impact de la drogue sur différentes histoires de pathogenèse
3. Polymorbidité - que homme âgé, plus de maladies des maladies en même temps
4. Besoin de corriger les effets indésirables des drogues
5. Réduire le nombre de techniques et introduction de médicaments (commodité pour le patient, économies de main-d'œuvre des agents de santé)
Types d'interaction:
JE.. Interaction pharmaceutique -Le type d'interaction associé à une réaction physicochimique entre les médicaments dans le processus de préparation de médicaments avant l'introduction de ces fonds dans le corps humain
A) Erreurs typiques menant à une incompatibilité pharmaceutique: la décharge de recettes complexes, un stockage incorrect, n'est pas pris en compte la possibilité d'adsorption de médicaments à la surface des plastiques (nitrates biologiques)
B) Problèmes liés à la perfusion: mélange de sels solubles, de dérivés d'acides faibles insolubles ou de bases entraînent leur précipitation; Les glycosides cardiaques et les alcaloïdes sont hydrolysés sous forme de dosage liquide; PH du milieu (alcaloïdes précipités de milieu alcalin)
C) Recommandations: 1) Tous les mélanges sont préférables de préparer EX Tempore 2) La solution la plus fiable avec un réseau local 3) Toutes les solutions doivent être vérifiées pour la présence de STRIPS 4) L'interaction peut se produire sans changement visible de solutions 5) n'est pas possible d'ajouter des médicaments au sang et aux solutions d'AK 6) en l'absence d'instructions spéciales, les médicaments doivent être dissous dans un glucose de 5% de R-RE-RE (PH 3.5-6.5), une solution isotonique NaCl (pH 4.5-7.0 ).
La solution de glucose, un HCl stabilisé, incompatible avec adrénaline, benzylpénicilline, apomorphine, canamicine, vitamine C, oléenne, glycosides cardiaques. Les glycosides cardiaques sont incompatibles avec l'atropine, la papaverine, la platinilline. AB est incompatible avec l'héparine, l'hydrocortisone. Les vitamines des groupes sont incompatibles les unes avec les autres, avec des vitamines PP, C. La vitamine RR et avec entre elles sont incompatibles.
Il est impossible de mélanger d'autres médicaments: phénothiazide, chlorpromazine, barbituriques, préparations de vitamine C, amphotéricine, furosémide, sunfadiazine, aminophylline, adrénomimétique.
II.. Pharmacologique- l'interaction des médicaments qui se manifeste uniquement dans le corps humain après leur utilisation commune
A) pharmacocinétique
1) à la phase d'aspiration.
Sous l'introductionPar. Système d'exploitation L'interaction est déterministe:
1. environnement d'acidité
2. Interaction directe dans le tractus gastro-intestinal
Les tétracyclines interagissent avec le calcium, l'aluminium, le fer, le magnésium pour former des complexes de chélate. La cholestyramine perturbe l'absorption des dérivés, du calcium, de la varvarina, de la digoxine, de la digitoxine, de vitamines solubles dans les graisses, de triméthopris, de la clindamycine, de la céphalexine, de la tétracycline. Les préparations de fer sont mieux absorbées par la vitamine C. Préparations de fer avec des carbonates, les tétracycles sont mal absorbés.
3. Motoric Zhkt.
Ralentissez les péristaltiques: certains antidépresseurs, drogues antihydamine, phénothiazine antipsychotique, stupéfiants, augmentent l'absorption de la digoxine, des corticostéroïdes, des anticoagulants, réduisent l'absorption de la lévodopa. Améliorer les péristaltiques et augmenter l'évacuation du tractus gastro-intestinal: métoclopramide, laxatifs. Réduire l'absorption des médicaments: Phénobarbital - Grrereofulvine, Aspirine - Indométhacine et Diclofenac, Pask - Rifampicine.
Façons de contrôler l'aspiration pendant l'administration parentérale: Anesthésiques locaux + adrénaline + phényléphrine - succion réduite de l'anesthésiques locaux
4. flore intestinale
5. Changer le mécanisme d'aspiration
2) Pendant la distribution et le dépôt:
1. Interaction directe dans le plasma sanguin: gentamicin + ampicilline ou carbénicilline - réduire l'activité de la gentamicine
2. Déplacement compétitif dû à l'albumine dans le plasma sanguin: indométhacine, digitoxine, warfarine sont associés à des protéines sanguines de 90 à 98%, par conséquent une augmentation de la fraction libre des médicaments deux fois - une forte augmentation des effets toxiques; Les AINS sont déplacées: warfarine, phénytoïne, méthotrexate.
Déterminations qui déterminent la signification clinique de cette interaction:
ü Valeur VD (Big - Pas de problèmes, petit - possible)
ü L'influence d'une substance LAN sur l'activité des mécanismes de transport par le biais des mécanismes d'autres médicaments: Augmente davantage le transport LS - Insuline, ACTH, Angiotensine, Kinines, etc. L'insuline augmente la concentration d'isoniazide uniquement dans les poumons et la concentration du coton n'est que dans le MMC.
3. Mouvement dû aux protéines de tissu: le comté déplace Digoxin + réduit l'excrétion rénale, de sorte que le risque de toxicité de la digoxine augmente
3) Dans le processus de métabolisme
LS peut augmenter ou réduire l'activité du cytochrome P450 et ses enzymes (l'éthanol augmente l'activité de certains isoenzymes de cytochrome)
Inhibiteurs des enzymes qui entrent souvent dans une interaction:
1. AB: Ciprofloxacine, Erythromycine, Isoniazide, Metronidazole
2. Préparations cardiovasculaires: Amiodaron, Diltiazem, Quinidine, Verapamil
3. Antidépresseurs: fluoxétine, Sernesen
4. Préparations antisecretartaires: Cimétidine, oméprazole
5. Anticipants: Allopurinol
6. Fungicides: fluconazole, intrakanazole, kétoconazole, miconazole
7. Antiviral: Indinavir, Retoeavir, Saquinavir
8. Autres: Disulfiram, sodium ValProat
Préparations qui donnent des effets toxiques lors de l'inhibition de la MAO: l'adréthomimétique, la sympathomimétique, anti-pharynésoniques, analgésiques narcotiques, phénothiazines, sédatifs, diurétiques antihypertenseurs, médicaments hypoglycémiques
4) Dans le processus de dérivation - Plus de 90% des HPS sont excrétés d'urine.
Effet sur le pH de l'urine et le degré d'ionisation des médicaments, sur leur lipophile et leur réabsorption
1. Interaction lors de la diffusion passive: une partie du médicament est excrétée inchangée, une partie de la LS est ionisée à un pH de l'urine de 4,6-8,2. L'appui de l'urine est cliniquement important: empoisonnement avec de l'acide acétylsalicylique ou phénobarbital, lors de la prise de sulfanimamides (risque réduit de cristallurie), la réception de la quinidine. Augmentation de l'urine Acidité: l'excrétion d'amphétamine augmente (il est pratique d'identifier ce médicament aux athlètes)
2. Interaction Dans la période de transport actif: Propensions + pénicilline Augmente la durée du mouvement de la pénicilline, probénéécurant + salicylates - Élimination de l'effet urdilaurique du probénécide, pénicilline + SA - Réduisant l'excrétion de la pénicilline
L'effet de la composition d'urine sur l'excrétion de LV:
Le sucre croissant dans l'urine est une augmentation de l'excrétion: vitamine C, chloramphénicol, morphine, isoiazide, glutathion et leurs métabolites.
B) pharmacodynamique - Il s'agit de l'interaction des médicaments associés à un changement de pharmacodynamique de l'un d'entre eux sous l'influence d'une autre (sous l'influence des hormones thyroïdiennes, la synthèse des adrénorécepteurs B dans le myocardium est améliorée et l'effet de l'adrénaline par myocarde est amélioré) .
Exemples d'interactions synergiques indésirables cliniquement significatives:
NSAID + Varvarin - une augmentation du risque de saignement
Alcool + benzodiazépines - la potentialisation de l'effet sédatif
Inhibiteurs de l'ACE + K + ---- SBERLING DIURETIQUES - Risque accru d'hypercalémie
Verapamil + B-adrenoblocers - Bradycardie et Asistolie
L'alcool est un forte inducteur d'enzymes microsomales, conduit au développement de la tolérance aux médicaments (en particulier pour l'anesthésie et les équipements de sommeil), augmente le risque de dépendance à la drogue.
43. L'interaction des médicaments. L'antagonisme, la synergie, leur point de vue. La nature des changements dans l'effet des médicaments (activité, efficacité), en fonction du type d'antagonisme.
Dans l'interaction des médicaments, il est possible de développer les États suivants: a) Renforcer les effets de la combinaison de médicaments LS B) affaiblissement des effets de la combinaison de la LS C) incompatibilité des médicaments
Le renforcement des effets de la combinaison HP est mis en œuvre en trois versions:
1) Sommation des effets ou d'une interaction additive - vue interaction médicinale Dans lequel l'effet de la combinaison est la somme simple des effets de chacun des LS séparément. C'est-à-dire 1+1=2 . Il est caractéristique de la drogue d'un groupe pharmacologique, qui comporte une cible commune d'actions (activité de météral acide de la combinaison d'hydroxyde d'aluminium et de magnésium est égale à la somme de leurs capacités de repas acide séparément)
2) La synergie est un type d'interaction dans lequel l'effet de la combinaison dépasse la somme des effets de chacune des substances prises séparément. C'est-à-dire 1+1=3 . La synergie peut concerner les effets désirés (thérapeutiques) et indésirables des médicaments. L'administration combinée des diurétiques thiazidiques du dichlatiacé et un inhibiteur de l'ACE Enalapril entraîne une augmentation de l'action hypotensive de chacun des moyens, qui est utilisé dans le traitement de AG. Cependant, le but simultané des antibiotiques aminoglycosidiques (gentamicine) et la diurétique de la boucle de furosémide provoque une forte augmentation du risque de surexy et de surdité.
3) Potentiation - Le type d'interaction médicamenteuse, dans lequel l'un des LS, qui n'a pas d'effet en soi, peut entraîner une forte augmentation de l'action d'un autre médicament. C'est-à-dire 1+0=3 (L'acide clawulanique n'a pas d'effet antimicrobien, mais est capable de renforcer l'effet de l'antibiotique antibiotique B-lactam en raison du fait qu'il bloque la b-lactamase; l'adrénaline n'a pas d'action essentielle locale, mais quand elle est ajoutée à un Solution d'ultraakine, il allonge considérablement son effet anesthésique en raison de l'anesthésie de décélération d'aspiration du site d'injection).
Effets d'affaiblissement HP quand ils sont co-utiliser appelé antagonisme:
1) Antagonisme chimique ou antidotétisme - réaction chimique des substances entre elles avec la formation de produits inactifs (antagoniste chimique de la déféronamine des ions de fer, qui les lie dans des complexes inactifs; sulfate de protamine, dont la molécule a une charge positive excessive - un antagoniste chimique de l'héparine, dont la molécule a une charge négative excédentaire). L'antagonisme chimique est basé sur des antidotes (antidote).
2) Antagonisme pharmacologique (droit) - antagonisme causé par une action multidirectionnelle de 2 substances médicinales sur les mêmes récepteurs des tissus. L'antagonisme pharmacologique peut être compétitif (réversible) et non concurrentiel (irréversible):
A) Antagonisme compétitif: l'antagoniste compétitif se lie de manière réversible au centre actif du récepteur, c'est-à-dire que l'action de l'agoniste. T. K. Le degré de liaison de la substance avec le récepteur est proportionnel à la concentration de cette substance, puis l'action de l'antagoniste compétitif peut être surmontée si vous augmentez la concentration d'agoniste. Il déplacera l'antagoniste du centre récepteur actif et provoque intégralement la réponse tissulaire. T. À propos. L'antagoniste compétitif ne change pas l'effet maximum de l'agoniste, mais il nécessite sa concentration plus élevée pour interagir avec le récepteur. L'antagoniste compétitif déplace la courbe "effet de dose" pour l'agoniste au droit par rapport aux valeurs initiales et augmente le CE50 pour l'agoniste sans affecter la valeur de la valeur de Max.
Dans la pratique médicale, l'antagonisme concurrentiel utilise souvent. Étant donné que l'effet d'un antagoniste compétitif peut être surmonté si sa concentration est tombée en dessous du niveau de l'agoniste, dans le traitement des antagonistes concurrentiels, il est nécessaire de maintenir constamment son niveau assez élevé. En d'autres termes, l'effet clinique de l'antagoniste concurrentiel dépendra de la période de sa semi-élimination et de la concentration d'agoniste complet.
B) Antagonisme non concurrentiel: un antagoniste non concurrentiel est associé presque irréversible avec le centre actif du récepteur ou interagit en général avec son centre de machines alto. Par conséquent, peu importe la concentration d'agoniste augmente - il n'est pas capable d'injecter un antagoniste d'un récepteur. Puisque, une partie des récepteurs, associée à un antagoniste non compétitif, n'est plus en mesure d'activer , Valeur E.Max Fréquence, l'affinité du récepteur à l'agoniste ne change pas, la valeur de l'EC50 reste donc identique. Sur la courbe de la courbe "dose-effet", l'effet d'un antagoniste non concurrentiel se manifeste sous la forme d'une compression de la courbe par rapport à l'axe vertical sans son déplacement à droite.
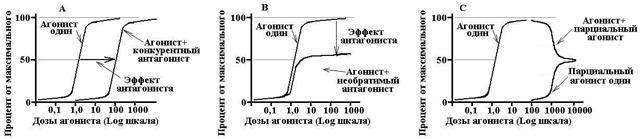
Schéma 9. Types d'antagonisme.
A - L'antagoniste concurrentiel transfère la courbe "dose-effet" à droite, c'est-à-dire réduisant la sensibilité du tissu à l'agoniste, sans changer son effet. B - L'antagoniste non concurrentiel réduit la valeur de la réponse tissulaire (effet), mais n'affecte pas sa sensibilité à l'agoniste. C - une option d'application d'un agoniste partiel à l'arrière-plan d'un agoniste complet. Au fur et à mesure que la concentration augmente, l'agoniste partiel déplace la pleine de récepteurs et finalement la réponse du tissu diminue de la réponse maximale à l'agoniste complet, à la réponse maximale à l'agoniste partiel.
Les antagonistes non compétitifs sont appliqués moins souvent dans la pratique médicale. D'une part, ils ont un avantage incontestable, car ils ne peuvent pas être surmontés après liaison au récepteur et ne dépendent donc pas de la période de semi-élimination de l'antagoniste ou du niveau d'agoniste dans le corps. L'effet d'un antagoniste non concurrentiel ne sera déterminé que par le taux de synthèse de nouveaux récepteurs. Mais d'autre part, si une surdose de ce médicament se produit, il sera extrêmement difficile d'éliminer son effet.
|
Antagoniste compétitif |
Antagoniste non concurrentiel |
|
Ressemble à un bâtiment sur l'agoniste |
Sur la structure diffère d'agoniste |
|
Associé au centre actif du récepteur |
Associé au récepteur de cellules alto |
|
Déplace la courbe "effet de dose" à droite |
Déplace la courbe "effet de dose" verticalement |
|
L'antagoniste réduit la sensibilité tissulaire à l'agoniste (EC50), mais n'affecte pas l'effet maximum (EMAX), qui peut être obtenu à une concentration plus élevée. |
L'antagoniste ne change pas la sensibilité du tissu à l'agoniste (EC50), mais réduit l'activité interne de l'agoniste et de la réponse maximale des tissus à celui-ci (EMAX). |
|
L'action antagoniste peut être éliminée par une forte dose d'agoniste |
L'action de l'antagoniste ne peut être éliminée par une dose élevée d'agoniste. |
|
L'effet de l'antagoniste dépend du ratio de doses agonistes et antagonistes |
L'effet de l'antagoniste ne dépend que de sa dose. |
L'antagoniste compétitif vers les récepteurs de l'angiotensine de l'angiotensine est Lozartan, il perturbe l'interaction de l'angiotensine II avec des récepteurs et contribue à réduire la pression artérielle. L'action du Losartan peut être surmontée si vous introduisez une dose élevée d'angiotensine II. Un antagoniste non concurrentiel concernant les mêmes récepteurs AT1 est Valsartan. Son action ne peut être surmontée même avec l'introduction de fortes doses d'angiotensine II.
Intéressant est l'interaction qui a lieu entre les agonistes complètes et partiels des récepteurs. Si la concentration de l'agoniste total dépasse le niveau de partielle, la réponse maximale est observée dans le tissu. Si le niveau de l'agoniste partiel commence à augmenter, il déplace l'agoniste complet due au récepteur et la réponse du tissu commence à diminuer du maximum pour un agoniste complet, au maximum pour un agoniste partiel (c'est-à-dire un tel niveau à qui prendra tous les récepteurs).
3) Antagonisme physiologique (indirect)- L'antagonisme associé à l'influence de 2 substances médicinales sur divers récepteurs (cibles) dans les tissus, ce qui entraîne un affaiblissement mutuel de leur effet. Par exemple, l'antagonisme physiologique est observé entre l'insuline et l'adrénaline. L'insuline active les récepteurs de l'insuline entraînant augmenter le transport de glucose dans la cellule et le niveau de glycémie diminue. L'adrénaline active B2-adrénorécepteurs du foie, des muscles squelettiques et stimule la carie glycogène, qui entraîne finalement une augmentation de la glycémie. Ce type d'antagonisme est souvent utilisé pour fournir des soins d'urgence aux patients atteints d'une surdose de l'insuline, ce qui a conduit à un coma hypoglycémique.
44. Effets secondaires et toxiques des substances médicinales. Effet teratogénique, embryotoxique et mutagène des médicaments.
Effets secondaires - les effets qui se produisent lorsque l'utilisation de substances de doses thérapeutiques et constituent le spectre de leur action pharmacologique (la morphine analgésique des doses thérapeutiques provoque l'euphorie), peut être primaire et secondaire:
A) Effets secondaires principaux - En conséquence directe de l'influence de ce médicament sur un certain substrat (hypospalivation lors de l'application de l'atropine pour éliminer le bradiatum)
B) Effets secondaires secondaires - Effets indésirables émergents indirectement (AB, supprimant une microflore normale, peut entraîner une superinfection)
Effets toxiques - des effets indésirables qui se manifestent dans cette HP lors de la sortie de la gamme thérapeutique (surdose LS)
La sélectivité de l'action LAN dépend de sa dose. Plus la dose de la drogue est élevée, moins il devient des élections.
Action tératogène- la capacité de la drogue lors de la nomination de sa femme enceinte de causer des anomalies anatomiques pour le développement du fœtus (Talidomide: Focomelia, Antoblatural LS: Multiples défauts)
Action embryotoxique - Effets indésirables qui ne sont pas associés à la violation de l'organogenèse au cours des trois premiers mois de la grossesse. Aux dernières échéances manifestes Action féthotoxique.
Effet mutagénique de HP - Dommages causés à la cellule germinale et à son appareil génétique de médicaments, qui se manifeste par un changement du génotype de progéniture (adrénaline, cytostatique).
Action cancérogène ls. - la capacité de certains ls à induire la carcinogenèse.
45. Aspects médicaux et sociaux de la lutte contre la toxicomanie, la toxicomanie et l'alcoolisme. Le concept de toxicomique.
« Le fait que l'humanité dans son ensemble se produise jamais sans paradis artificiel, improbable. La plupart des hommes et des femmes mènent une vie aussi douloureuse que meilleur cas alors monotorne, misérable et limité que le désir de "partir" d'elle, de se déconnecter au moins quelques instants, et il y a toujours eu l'un des principauxSouhaiter Âmes"(Huxley, le travail de" les portes de la perception "))
1) La toxicomanie - l'état de la psyché et / ou de la condition physique, qui est une conséquence de l'impact sur le corps du réseau local et se caractérise par des réactions comportementales spécifiques, difficiles à re-prendre la LS pour obtenir un effet mental spécial ou éviter l'inconfort dans l'absence de HC dans le corps. La toxicomanie est caractérisée par:
MAIS) Dépendance psychologique - Développement de détresse émotionnelle lors de l'arrêt de la réception des médicaments. Une personne se sent vide, immergée dans la dépression, sentant un sentiment de peur, d'anxiété, son comportement devient agressif. Tous ces symptômes psychopathologiques se posent contre le fond des réflexions sur la nécessité de se présenter un LS, qui a provoqué une dépendance. Le désir de recevoir des médicaments peut varier d'un désir simple à la soif passionnée d'admission de drogues, qui absorbe tous les autres besoins et se transforme en sens de la vie humaine. On pense que la dépendance psychologique se développe lorsqu'une personne apparaît une conscience que le bien-être optimal peut atteindre exclusivement grâce à l'introduction de HP. La base de la dépendance psychologique - la foi d'une personne en vigueur (dans la littérature, des cas de développement de la dépendance psychologique sur placebo).
B) Dépendance physique - violation de l'état physiologique normal de l'organisme, qui nécessite une présence constante de médicaments pour maintenir l'état d'équilibre physiologique. L'arrêt du médicament est causé par le développement d'un complexe de symptômes spécifique - le syndrome abstineent - un ensemble de troubles mentales et neurovétatives sous la forme d'une fonction de la fonction sur le côté opposé à celui qui est caractéristique de l'action ( La morphine élimine la douleur, opprime le centre respiratoire, rétrécit les élèves, provoque une constipation; lors de l'abstinence chez le patient, il y a des douleurs douloureuses de constipation, une respiration bruyante fréquente, des élèves sont élargis et une diarrhée obstinée se développe)
DANS) Tolérance. La tolérance aux moyens de causer une dépendance à la drogue est souvent transversale, c'est-à-dire que, il se produit non seulement à ce composé chimique, mais également à tous les composés structurellement similaires. Par exemple, chez les patients atteints de dépendance médicinale à la morphine, la tolérance se produit non seulement, mais également à d'autres analgésiques opioïdes.
Pour le développement de la toxicomanie, la présence de 3 critères n'est pas une condition préalable, dans le tableau 3 présente les principaux types de drogues et composants de composants.
Les opioïdes, les barbituriques, l'alcool causent de graves dépendances physiques, psychologiques et de tolérance. Anxiolitiques (diazépam, alprazolam) causer de préférence une dépendance psychologique.
2) Dépendance à la drogue (dépendance narcotique)- Il s'agit d'une forme extrêmement sévère de toxicomanie, de l'utilisation compulsif des médicaments, caractérisée par une imposition croissante et irrésistible à l'introduction de ce médicament, augmentant ainsi sa dose. La compulsivité de l'attraction signifie que la nécessité de l'introduction de médicaments domine le patient sur tous les autres (même vitaux). Du point de vue de cette définition, l'attrait de la dépendance à la morphine - drogue, tandis que l'attraction de la nicotine est une dépendance à la drogue.
3) La toxicomanie - caractérise une attraction moins intense pour la réception de médicaments, lorsque le rejet du médicament ne provoque que un sentiment d'inconfort léger, sans le développement de la dépendance physique ou une image détaillée de la dépendance psychologique. T. À propos. La dépendance couvre cette partie de la dépendance à la drogue qui ne relève pas de la définition de la toxicomanie. Par exemple, la toxicomanie mentionnée ci-dessus est la nicotine - une forme de dépendance.
4) Abus de drogue - Utilisation non autorisée de médicaments dans de telles doses et de telles manières qui diffèrent des normes médicales ou sociales adoptées dans cette culture et à cette époque. T. À propos. L'abus ne couvre que des aspects sociaux de la consommation de drogues. Un exemple d'abus est l'utilisation de stéroïdes anabolisants dans des sports ou d'améliorer le physique des jeunes hommes.
5) Alcoolisme - Abus d'alcool chronique (alcool éthylique), menant de temps à des dommages à un certain nombre d'organes (hépatique, tractus gastro-intestinal, SNC, système cardiovasculaire, système immunitaire) et accompagné d'une dépendance psycho-physique.
6) Toxicomie - Abus chronique de divers LS (y compris des médicaments, de l'alcool, des hallucinogènes), manifesté par divers troubles mentaux et somatiques, violation du comportement, dégradation sociale.
Traitement médicinal Tâche dure et ingrat. La technique effective n'a pas encore été créée, ce qui garantirait le succès du traitement de plus de 30 à 40% des patients. La réalisation de tous les résultats notables n'est possible qu'avec la coopération totale des efforts du patient, un médecin et l'environnement social dans lequel les malades (principe de volontarisme et d'individualité). La base des techniques modernes est les principes suivants:
ü Méthodes psychothérapeutiques et d'emploi;
ü Traitement et réhabilitation de groupe (Sociétés d'alcooliques anonymes, toxicomanes)
ü Abolition progressive ou forte du médicament au milieu de la therapie de désintoxication
ü Traitement de substitution (remplacement de la drogue narcotique lentement et des analogues durables avec leur annulation ultérieure; par exemple, t. n. N. Le programme de la méthadone substitution thérapie dans les toxicomanes d'héroïne)
ü Traitement avec des antagonistes spécifiques (naloxone et naltrexon) ou des moyens de sensibilisation (Teturam)
ü Méthodes neurochirurgicales de cryodétructuration de la courroie de pain d'épice et d'hippocampe
47. Types de pharmacothérapie. Problèmes déontologiques de la pharmacothérapie.
Pharmacothérapie (FT.) - Un ensemble de méthodes de traitement basées sur l'utilisation de médicaments. Les principaux types de FT:
1. Etiotrope FT - Correction et élimination des causes de la maladie (AB dans des maladies infectieuses)
2. Pathogenetic FT - Impact sur le mécanisme du développement des maladies (inhibiteurs de l'ACE à AG)
3. FT symptomatique - Élimination des symptômes de la maladie lorsqu'il est impossible d'influencer sa cause ou sa pathogenèse (AINS de grippe)
4. Remplacement FT - L'utilisation de médicaments avec insuffisance de BAV naturel (insuline à la SD)
5. FT préventif (vaccin, sérum, acide acétylsalicylique avec IBS)
L'attitude de la société à la drogue sur scène moderne : 1) désir d'obtenir des avantages sans risque 2) L'espoir d'un miracle, de super-pli 3) malentendu du risque d'utilisation de la LS 4) scandale et "indignation juste", estimation hâtive de la LS 5) Le désir de recevoir de nouveaux ls
L'attitude du médecin envers LS: Optimisme thérapeutique (espoir la LS, en tant que composant puissant de la thérapie), nihilisme thérapeutique (déni de nouveaux médicaments, engagement à certains médicaments, méfiance du nouveau réseau local)
Conformité (engagement) patient pour le traitement: 1) Comprendre les instructions des objectifs du médecin et du traitement 2) Le désir de suivre avec précision les prescriptions du médecin.
Actuellement dans le monde d'environ 100 000 ch, plus de 4 000 personnes sont enregistrées en République de Biélorussie, environ 300 médicaments sont essentiels. L'étude de la pharmacologie contribue à ne pas se noyer dans la mer de la drogue.
48. Principes de base du traitement et de la prévention de l'intoxication médicinale. Antidote thérapie.
Classification des substances d'empoisonnement:
1. En appartenant à certaines classes de composés chimiques: barbituriques, benzodiazépines, cyanides.
2. D'origine: Nébiologie (acide, alcalin, sels de métaux lourds), produits toxiques d'activité vitale de certains MB (botulinum), origine végétale (alcohyroïdes, glycosides), origine animale (serpents, abeilles)
3. En degré de toxicité: a) extrêmement toxique (DL50< 1 мг/кг) б) высоко токсические (1-50) в) сильно токсические (50-500) г) умеренно токсические (500-5000) д) мало токсические (5000-15000) е) практически нетоксические (> 15.000)
4. Selon des mesures toxicologiques: a) Neuro-paralytique (bronchospasme, hachage) B) Skin-résorbatif c) général-toxique (crampes hypoxiques, coma, paralysie) d) étouffement d) étouffement d) larme et ennuyant e) psychotrope (violation du mental activité, conscience)
5. Selon la sphère d'utilisation préférentielle: poisons industriels, pesticides, poisons ménagers, substances d'intoxication combattantes, substances médicinales.
6. En fonction de la toxicité du médicament: la liste des médicaments, la nomination, l'utilisation, le dosage et le stockage de ceux dus à une toxicité élevée doivent être effectués avec une prudence élevée. La même liste comprend les médicaments causant une toxicomanie; Liste de B - HP, but, application, dosage et stockage de ce qui doit être effectué par des précautions en raison de complications possibles lorsqu'elles sont appliquées sans contrôle médical.
L'effet toxique sélectivement des médicaments.
A) Cardiotoxic: glycosides cardiaques, préparations de potassium, antidépresseurs
B) Neurotoxique: agents psychopharmacologiques, oxychinolines, aminoglycosides
C) hépatotoxique: tétracyclines, lévomycétine, érythromycine, paracétamol
D) néphrotoxique: vancomycine, aminoglycosides, sulfonamides
E) gastroenterotoxique: fonds anti-inflammatoires de stéroïdes, AINS, Reserpine
E) hématotoxique: cytostatique, lévomycétine, sulfonamides, nitrates, nitrites
G) pneumotoxique
Toxicocinétique - étudie l'aspiration, la distribution, le métabolisme et l'élimination des médicaments adoptés dans des doses toxiques.
L'apport de substances d'empoisonnement dans le corps est possible a) entéral B) parentérale. La rapidité et l'exhaustivité de l'aspiration reflètent le taux de développement de l'effet toxique et de sa gravité.
Distribution dans le corps: VD \u003d D / CMAX est un volume valide dans lequel une substance d'empoisonnement est distribuée dans le corps. VD\u003e 5-10 L / kg est difficile à enlever (antidépresseurs, phénothiazines). Vd.< 1 л/кг – ОВ легче удалить из организма (теофиллин, салицилаты, фенобарбитал).
Surdose - Modifications des processus pharmacocinétiques: solubilité, communications avec des protéines, métabolisme ® Augmentation significative de la fraction libre Effet toxique LS ®.
La cinétique de premier ordre avec une augmentation de la concentration de LS passe dans la cinétique de commande zéro.
Stage toxigène - Thérapie désinfectante, étape somatogène - thérapie symptomatique.
Toxicodynamique . Mécanismes de base toxiques:
A) Médiateur: droit (selon le type de blocus compétitif - Fos, psychomimétiques) et indirects (activateurs ou inhibiteurs d'enzymes)
B) Interaction avec les biomolécules et les structures intracellulaires (substances hémolytiques)
C) Métabolisme par type de synthèse mortelle (alcool éthylique, thiophos)
D) Enzyme (poisons serpents, etc.)
Types d'action: local, réflexe, résorbatif.
Classification de l'empoisonnement:
1. Éthiopathogénétique:
a) aléatoire (auto-traitement, erreur)
b) délibéré (avec le but de suicide, meurtre, développement de l'état impuissant affecté)
2. Clinique:
a) Selon le taux de développement d'intoxication: aigu (admission une fois ou avec un intervalle de dose toxique de substance de substance), subaiguë (développement de ralentissement d'une image clinique après une admission ponctuelle), chronique
b) En fonction de la manifestation du syndrome principal: la défaite de la Cass, la défaite de DS, etc.
c) En fonction de la gravité de l'état du patient: lumière, moyenne, sévère, extrêmement lourde
3. Nonzologique: prend en compte le nom de la LS, le nom du groupe de substances
Le mécanisme de décès global d'intoxication:
A) la défaite du CSS:
1) La pression artérielle réduite, l'hypovolémie des navires périphériques, l'effondrement, le brady - ou la tachycardie (antidépresseurs tricycliques, bloquants bêta, bloquants de calcium)
2) Arythmies (tachycardie ventriculaire, fibrillation - antidépresseurs tricycliques, théophylline, amphétamine)
B) défaite du système nerveux central: stupeur, dépression respiratoire Koma ® (médicaments, barbituriques, alcool, hypno-sédatif LS)
C) Crampes, hyperréactivité musculaire et rigidité ® Hyperthermie, Mioglobinurie, insuffisance rénale, hypercalémie
Triade toxicologique:
1) L'historique de la durée de l'application, de la dose et de la substance ®.
2) Évaluation de l'état de conscience dans les symptômes: respiration, pression artérielle, température corporelle
3) Données de laboratoire
Principes de base du traitement:
JE. Première aide d'urgence: Souffle artificiel, massage cardiaque, thérapie anti-lente, contrôle de l'équilibre de l'eau et de l'électricité
II.. Le délai d'aspiration et l'élimination du corps ne sont pas tentés:
But: arrêter le contact avec
1. Chemin parentérale:
a) à travers les poumons:
1) arrêter l'inhalation
2) Substances irritantes (alcool amponique, formaldéhyde) ® Fixez les mouvements actifs, au chaud, donnez de l'oxygène et des séchoirs (dans l'alcool ammonique, un vinaigre de dégoamer et le formaldéhyde est une solution diluée d'alcool ammoniac)
b) à travers la peau: rincer à une quantité abondante d'eau tiède avec du savon ou du détergent, un antidote spécifique, une neutralisation et une terminaison d'exposition à la peau sur la peau (FOS: laver à l'eau, enlevé avec 10-15% de l'alcool amponique ou 5-6% de solution d'hydrocarbonate de sodium avec de l'eau; phénolzol: huile végétale ou éthylène glycol, mais d'huile de vaseline impossible, de KMNO4: 0.5-1% de solution d'acide ascorbique ou de volumes égaux de 3% de peroxyde d'hydrogène et 3% de solution d'acide acétique, CCL4 , Turpentine, essence: eau de savon chaud)
c) Lorsque des injections dans la limbe: harnais au-dessus du site d'injection
d) En entrant dans l'oeil: lavage avec une solution saline ou du lait chaud pendant 10 à 20 minutes, pour creuser un anesthésique local; Si les acides et les alcalis, il est impossible de neutraliser. La consultation d'un ophtalmologiste est requise.
2. Chemin entéral: libérer l'estomac de Oh, accélérer le passage
a) élimination de s:
1) Pre-Prendre de l'eau. Il est impossible de prendre du lait (exception - des substances d'intoxication caustiques) et de l'éthanol (exclusion -metthanol).
2) Vomissements - est présenté principalement en cas d'empoisonnement avec de grandes pilules ou de capsules qui ne peuvent pas passer à travers la sonde. Vous pouvez provoquer réflexivement ou vomi (NaCl: 1 cuillère à soupe pour 1 tasse d'eau; Sirop d'hyquaan: adulte 2 cuillères à soupe, enfants 2 cuillères à café; moutarde: 1-2 cuillères à café sur un verre d'eau; Apomorphine: 5-10 mg / kg sous-cutané sauf les enfants de moins de 5 ans). Il est impossible de provoquer des vomissements après la réception: solvants biologiques - Le danger des inhalations, des détergents - Mousse, convulsives RY - Le danger de l'aspiration, des substances caustiques - des dégâts œsophagiens)
3) Protéger l'estomac - est un événement d'urgence et obligatoire. L'estomac est lavé, sinon plus de 4 à 6 heures passa du moment d'empoisonnement, parfois jusqu'à 10 heures; En empoisonnant avec de l'acide acétylsalicylique - après 24 heures. Pré-intuber le patient avec un tuyau avec un brassard gonflable: dans un état comateux en l'absence d'une toux et d'un réflexe laryngé. L'estomac est lavé à l'eau ou à 50 ° C physiathique, l'heure de la procédure est de 4 heures ou plus. À la fin du charbon activé par lavage et du sulfate de sodium.
b) Réduction de l'aspiration de l'aspiration gastro-intestinale: carbone activé à l'intérieur après la vidange de l'estomac + du sulfate de sodium ou du magnésium. Caractéristiques des mesures pour réduire l'aspiration:
1) solvants organiques: il est impossible d'induire des vomissements, de laver l'estomac après l'intubation, du carbone actif + de l'huile de vaseline
2) Détergents: il est impossible de provoquer des vomissements et de rincer l'estomac, il est nécessaire de donner beaucoup d'eau + de dégoamers (symétiques)
3) Acides et alcalis: Il est impossible de provoquer des vomissements, de laver l'estomac à travers la sonde, lubrifiée à l'huile de légumes après l'administration de l'analgésique narcotique - le seul témoignage du lait de don. En empoisonnement acide - antiacides, avec empoisonnement alcalin - acide citrique ou acétique.
III. Retrait du corps d'un bain de chaux
a) Dirésis forcés (conditions: flux sanguin rénal suffisant et filtrage glomère; en 24 heures pour verser 20-25 litres)
b) hémodialyse péritonéale
c) Hémosorption
d) Transfusion d'échanges de sang
e) hyperventilation forcée
Iv. Thérapie symptomatique des troubles fonctionnels.
Antidots: 1) Toxicotropique - Reliure, neutralisation et prévention de l'aspiration S: Valable sur le principe charbon activéAgir sur le principe chimique (Unitéiol, la pénicillamine, la pentazine)
2) toxicocinétique - accélérer la biotransformation OB (bromure trimécédoxy, thiosulfate de sodium, éthanol, jsc)
3) Pharmacologique - Atropine, Naloxon
4) antidote immunologique
Unitiol, succifère - lie les métaux lourds, les métalloïdes, les glycosides coeur. Esmolol lie la théophylline, la caféine. Calcium Trinatry Penotatat - Forme complexes avec des métaux de deux et trivalents.
49. Recette et sa structure. Règles générales Écriture de la recette. Réglementation de l'État du règlement des médicaments de prescription et de vacances.
Recette - cet appel de docteur écrit au pharmacien exigeant libérer le médicament dans une certaine forme et posologie indiquant la méthode de son utilisation
La recette distingue les parties suivantes:
1. INScriptio - titre, inscription. Voici la date du numéro de la recette, du nom de famille, des initiales et de l'âge du patient, du nom de famille et des initiales du médecin.
2. Invocatio - Appel au pharmacien. Il est exprimé par le mot "recette" (prise) ou désignation abrégée (RP)
3. Designatio Materiarum est la désignation ou le nom des médicaments avec l'indication de leurs doses. DANS recette complexe Le transfert de substances médicinales est effectué dans une séquence spécifique. Le premier indique la substance médicinale de base (base). Ensuite, écrivez des substances auxiliaires (adjuvuises). Après cela, indiquez les ingradients qui corrigent le goût, l'odeur, la couleur du médicament (corrigènes). Ces derniers sont des substances écrites qui donnent des médicaments à une certaine DOSSTItuences.
4. Subscriptio - Prescription (indication) Pharmacien. Cela indique une forme posologique, des opérations pharmaceutiques nécessaires à sa fabrication, le nombre de doses de médicaments libérées.
5. Signatura est une indication du patient sur la manière d'utiliser le médicament.
6. Subscriptio Medici - La signature du médecin qui a écrit la recette, son sceau personnel.
Appel du médecin au pharmacien, le nom des médicaments inclus dans la recette, le nom formulaire de dosage et la nature des opérations pharmaceutiques est écrite sur latin. Nom des médicaments, noms botaniques des plantes sont écrits avec lettre capitale. Le patient sur ordonnance est écrit en langues russes ou nationales.
Règles de nutrition de recette générale:
1. La recette est écrite sur une forme spéciale en fonction de l'écriture transparente, de l'encre ou du balle à bille Sans corrections.
2. La recette indique le nombre, le mois, l'année, le nom de famille, le nom, le patronyme et l'âge du patient, le nom de famille, le nom et le patronymique du médecin. Ensuite, le texte de la recette, où les noms inclus dans les noms de recette sont répertoriés dans le boîtier parental, indiquant leur quantité.
3. Unité de masse dans les recettes - gramme ou unités.
4. Si vous dépassez la dose maximale de substances toxiques et puissantes, elle est confirmée par inapplication
5. La recette est confirmée par la signature et le sceau personnel du médecin
Le RB exploite la réglementation de l'État des règles de prescription et de congé de drogue.
50. Règles d'écriture de fonds toxiques, stupéfiants et puissants.
La liste et les médicaments attribués, la nomination, l'utilisation, le dosage et le stockage de ceux dus à une toxicité élevée doivent être effectués avec une prudence élevée. La même liste comprend des médicaments qui causent une dépendance à la drogue.
La liste des médicaments attribués, de nomination, d'application, de dosage et de stockage doit être effectué par des précautions liées à des complications éventuelles lorsqu'elles sont appliquées sans contrôle médical.
Pour les médicaments toxiques et puissants, des doses maximales les plus élevées et les doses quotidiennes sont installées. Ces doses sont conçues pour les adultes atteintes de 25 ans. Lors de la recalculition des doses pour les personnes de plus de 60 ans sensibilité à l'âge à différents groupes Hp. Les doses de préparations, déprimantes CNS, ainsi que des glycosides cardiaques et des diurétiques diminuent de 50%, des doses d'autres médicaments toxiques et puissantes sont réduites à 2/3 de la dose d'un adulte. Les doses d'AB, de sulfonamides et de vitamines sont généralement les mêmes pour tous les groupes d'âgeà partir de 25 ans.
1. Les stupéfiants (liste A) sont émis sur prescription blanco Formulaires 2. Un blanc est un médicament. Doit être: la signature et l'impression du médecin traitant, la signature du médecin en chef du LPU, l'impression ronde du LPU.
2. Drogues toxiques (liste A), Potent (liste B) sont déchargées sur une forme de formulation 1. Il doit y avoir une signature et une impression personnelle du médecin, le sceau du LPU.
51. Médicaments sous contrôle. Médicaments interdits à écrire dans les recettes.
Les contrôles sont narcotiques, toxiques et puissants (voir. 20)
A) LS, non enregistré en République de Biélorussie et non autorisé à appliquer
B) HP à la demande des patients et de leurs proches sans inspection du patient et du diagnostic
C) Recettes pour les stupéfiants pour les injections, éther anesthésieux, chloroéthyle, pentamine, fluorotan, oxybutérisme de sodium dans des ampoules, oxybutyrate de lithium, sulfate de baryum pour rayons X.
52. Modèles pharmacocinétiques (une chambre à chambre et deux chambres), des lois quantitatives d'aspiration et d'élimination des médicaments.
![]() Modèle à chambre unique.
Modèle à chambre unique.
L'organisme entier est un seul conteneur homogène. Hypothèses:
1) Le développement dynamique rapide est établi entre le contenu du médicament dans le sang et sa concentration dans les tissus extravasculaires
2) LS rapidement et uniformément réparties dans tout le volume sanguin
3) L'élimination des médicaments obéit la cinétique de premier ordre: le taux de réduction du contenu du médicament dans le sang est proportionnel à sa concentration
Si les mécanismes d'élimination du médicament (la biotransformation dans le foie, la sécrétion rénale) ne sont pas saturés de l'introduction de la dose thérapeutique, le calendrier de connexion pour changer la concentration plasmatique dans le temps sera Ornée.
Inclinaison Axe lognomal - Kel, où Kel est un taux d'élimination constant et a la dimension du temps-1. La valeur de C0 est obtenue par extrapolation du graphique avant l'intersection avec l'axe d'ordonnée. Concentration plasmatique de LV (CT) à tout moment t après l'administration au corps est:
Ln ct \u003d ln c0 - kt. Élimination constante Kel, VD et autorisation générale (CL) sont liées à l'expression: cl \u003d k × vd
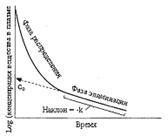 Modèle à deux chambres.
Modèle à deux chambres.
Souvent, après l'admission de médicaments dans le corps, il n'est pas possible d'atteindre rapidement l'équilibre entre la teneur en LS dans le sang et sa concentration dans le fluide extravasculaire. Ensuite, on pense que dans l'ensemble des tissus et des fluides biologiques du corps, deux caméras peuvent être distinguées, qui se distinguent par le degré d'accessibilité pour la pénétration de LS. La chambre centrale comprend du sang (souvent avec des organes intangiables - foie, reins), au fluide interstitiel périphérique les organes internes et les tissus.
L'horaire résultant montre l'initiale Phase de distribution (Le temps requis par le réseau local pour atteindre l'état d'équilibre entre les caméras centrales et périphériques et plus lentement plus lentement Phase d'éliminationPremier ordre.
La valeur C0 est obtenue par extrapolation Phases d'élimination Avant intersection avec le propriétaire de l'ordonnée. C0 est utilisé pour calculer le volume de distribution et la constante d'élimination. Les formules pour calculer CT et CL, données pour un modèle à chambre unique, sont également utilisées pendant la phase d'élimination des médicaments qui répondent aux conditions du modèle avec deux caméras.
53. Sélectivité et spécificité des médicaments. Les effets thérapeutiques, latéraux et toxiques de la drogue, leur nature du point de vue du concept de récepteurs. Stratégie thérapeutique de lutte contre les effets secondaires et toxiques de la drogue.
Spécificité - Ceci est lorsque la LS se lie à un type de récepteur strictement spécifique.
Sélectivité - Ceci est lorsque le médicament est capable de contacter un ou plusieurs types de récepteurs plus précisément que les autres.
Il est plus préférable d'utiliser la sélectivité du terme, car il est peu probable que toute molécule LS ne puisse être mise en contact avec un type de molécules de récepteur, car le nombre de récepteurs potentiels chez chaque patient a une valeur astronomique.
Action thérapeutique - L'effet pharmacologique principal souhaité, attendu de ce médicament pharmacologique.
Effets secondaires- les effets qui se produisent avec l'utilisation de substances de doses thérapeutiques et constituent le spectre de leur action pharmacologique.
Effets toxiques - des effets indésirables qui se manifestent dans cette HP lorsqu'ils quittent la gamme thérapeutique.
Communication d'effets thérapeutiques et toxiques des médicaments sur la base de l'analyse des mécanismes de récepteur et d'effecteur:
1) les effets thérapeutiques et toxiques médiés par le même mécanisme de récepteur-effecteur (la prazosine agit comme un antagoniste alpha-sélectif pour les récepteurs des navires GMC et a un effet hypotenseur dans l'hypertension essentielle, mais au cours de sa grande dose, un patient peut avoir une hypotension posturale)
2) Effets thérapeutiques et toxiques médiés par des récepteurs identiques, mais par divers tissus ou de divers chemins effecteurs (glycosides coeur servent à augmenter la capacité contractile du myocarde, en même temps, ils violent la fonction de digestion, vue en raison du blocus de Na + / K + membrane cellulaire)
3) Effets thérapeutiques et toxiques médiés par différents types de récepteurs (par exemple, la norépinéphrine a un effet hypertenseur à travers A1-AP, mais il provoque la tachycardie via B1-AP)
Stratégie thérapeutique de lutte contre les effets thérapeutiques et secondaires des médicaments:
1. LAN doit toujours être administré dans la dose la plus basse qui provoque un effet thérapeutique acceptable.
2. Réduire la dose d'une drogue en raison de l'objet de l'autre réseau local avec un effet similaire, mais à travers d'autres récepteurs et avec un profil de toxicité différent.
3. La sélectivité de l'action LS peut être augmentée en contrôlant la concentration de médicaments dans la zone des récepteurs de diverses parties du corps (utilisation locale de l'utilisation de LS-inhalation du salbutamol avec asthme bronchique)